常见问题
在这个页面汇总了常见的🙋系统使用问题,分析问题答疑等。您可以按下 CTRL + F 输入关键字进行搜索。
1. 系统使用
系统使用
Q:如何调整图片样品顺序?如何修改样品名?如何修改分组名?
A:新建分析时,样品名称、样品所在的分组,分组名称,样品的排序就已经按照您的设定确定了下来。系统是不支持对单个分析结果的上述信息进行修改的,如您需要修改这些信息需要新建一个新的分析,然后这些修改会应用到所有的分析点中。
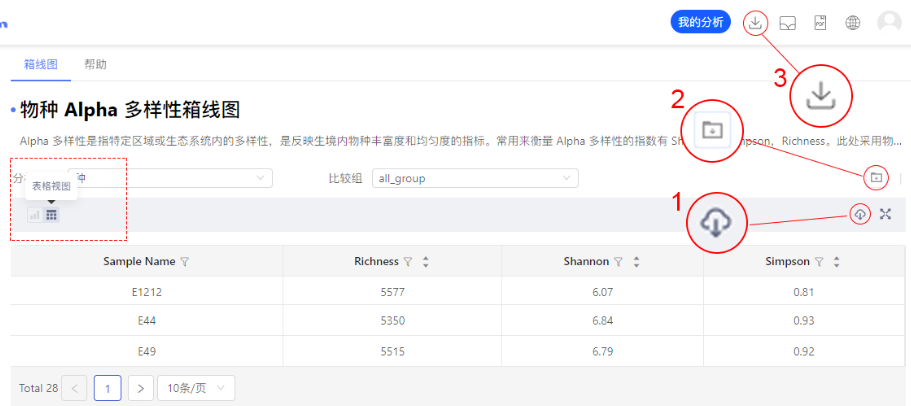
Q:分析结果下载?
A:系统提供了三种分析结果下载。您可以在结果页面图表切换 ① 处下载直接绘图数据,在② 处下载分析相关的中间数据(如果有),在 ③ 处下载整个分析报告的所有相关文件。
相关信息
分析报告中包括物种丰度表,但不包含功能丰度表。功能丰度表随 Clean Data 一起交付。
Q:结果文件夹中,不同文件的含义?
A:在结果目录中,files.txt 文件描述了文件夹和结果文件的含义。
Q:用 Excel 打开某些文件,发现文件内容,没有规律,每一行的数据很奇怪,是怎么回事?
A:测序数据的原始文件都很大,而做了一定的分析,包含所需信息的中间文件,也可能很大,使用 Excel 等软件打开,可能会有乱码或分行混乱等问题。例如,Kegg 的注释文件,直接使用 Excel 打开可能出现一行文本被分割为多行,如下图: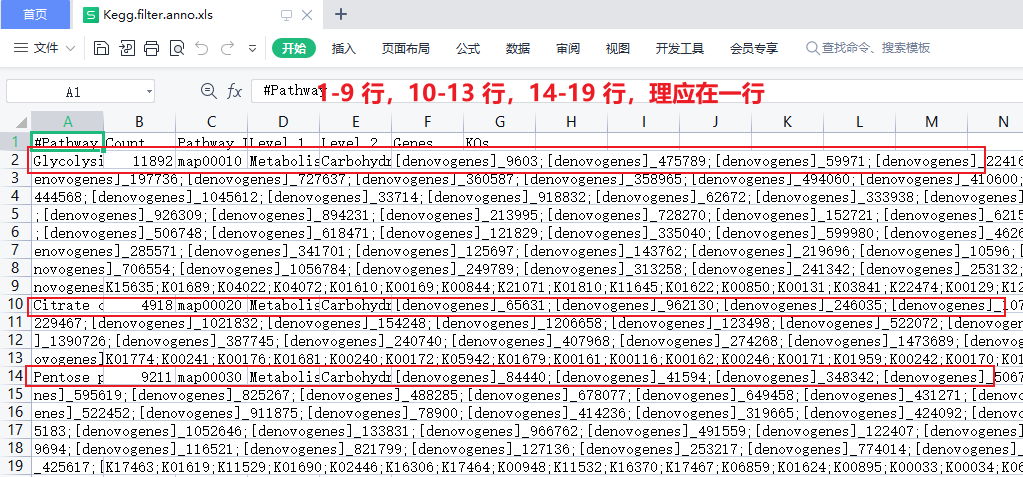
这种情况下,您可以采用以下方式查看文件:
- 在 Windows 下,使用文本编辑器查看相关文件。
- 使用 linux 系统的命令行工具查看相关文件。
Q:图片含义和图片解析?
A:我们为每个分析结果提供了详细的说明你可以在三个地方找到对应的说明
- 结果页面上方有分析的简短说明
- 在图片下方的说明中,有图片中元素的意义及文件基本意义
- 在分析点的帮助页面有详细的分析说明
Q:怎样对图片二次编辑和转换?
A:Dr.Tom 系统提供了常用的图片修改参数,您可以在系统中修改这么参数以使图片达到满意的状态。同时系统还提供了 PNG、SVG、PDF、JPG 格式图片的下载,若您需要更加个性化的图片编辑,您可以下载 SVG 或 PDF 格式的矢量图,然后使用合适的软件进行修改。
- SVG 格式图片文件可使用 svgdeveloper 软件进行修改,点击图中元素进行修改即可
- PDF 格式文件使用 AdobeIllustrator(AI) 继续修改; 图片转换: 利用上述软件打开图片后另存为需要的图片格式即可
Q:需要系统以外的其他个性化分析和可视化绘图服务?
A:推荐您使用华大旗下以科研文章级绘图为导向的数据可视化平台 Omiscribe(https://omiscribe.bgi.com/)。
项目次数问题
Q:需要新建分析,但次数已用完?
A:对于每一个项目我们默认提供了 3 次免费分析,如果您使用完这 3 次分析,需要您联系销售购买分析次数。
2. 湿实验和测序
湿实验
Q:宏基因组项目对样品重复有什么要求么?
A:宏基因组项目中每个分组组内的样品数量大于等于 3,推荐数量是 10。
Q:DNA 提取的实验方法、试剂?
A:提取完整的 DNA 是保证宏基因组结果质量的前提,然而环境样品组成复杂,当前并不存在一种适用所有类型样本的提取方案。以下列出较通用的提取步骤:
- 准备 5 块 96 孔深孔板,分别加入 600μL Buffer 磁珠结合液+20μL 蛋白酶 K+5μL RNase
A、700μL Wash 1、700μL Wash 2、700μL Wash 3、100μL Elution Buffer; - 取 100-200mg 样品至装有研磨珠的离心管中,加入 1mL Buffer ATL/PVP-10,至于高速研磨仪上研磨样品后至于 65℃ 孵育裂解 20min;
- 14000g 离心 5mim;将上清转移至新的离心管中,加入 0.6 mL Buffer PCI,旋涡混匀 15S;
- 12000rpm 离心 10min,将上清转移至加有磁珠结合液的深孔板中;
- 启动 Kingfisher 选择对应程序,将各个深孔板放于仪器对应位置,运行程序;
- 程序结束后,将 Elution Buffer 深孔板中 DNA 溶液转移至 1.5 mL 离心管中保存。
Q:样品被污染?如果存在较大的宿主的污染,且没有宿主基因组的参考序列可以进行宏基因组测序吗?
A:样品污染指的是测序结果中的非目标序列片段,除了采样和实验过程中引入的非目标细菌/真菌污染,宏基因组的污染还来自宿主。如果宿主的基因组序列在环境DNA中的量比较多,测序之后,我们没有办法通过已知的宿主基因组的序列来去污染,会对最后的分析结果造成很大影响,而且可用的数据量会很少;但是如果在提取的过程,宿主基因组的污染的量很少,后期的数据分析还是可用的,但是会存在一定的风险。因此应该在 DNA 提取过程中尽量避免来自宿主的 DNA。
测序
Q:质量评价 Q30、Q20 的含义?
A: 当测序仪检测到一个荧光信号,软件根据预设的规则把它判读成某个碱基,判错的概率为 ,则该碱基的质量分值(QualityValue,QV)定义为
当正确率为 99.9%时, 错误率 = 0.001,此时质量值
该碱基的质量也称为 Q30,类似的 Q20 表示碱基错误率为 1%,即准确率为 99%,Q10 表示碱基错误率为10%。在测序中,常用质量为 Q30、Q20 的碱基含量表示测序结果的整体质量,Q20 、Q30碱基的占比越高测序结果质量越高。
Q:什么是测序深度和覆盖度?
A:测序深度(Sequencing Depth)指的是测序得到的碱基总量(bp)与基因组(转录组或测序目标区域大小)的比值,是评价测序量的指标之一。测序深度计算公式为
:测序深度;:read 长度;:read 数量;:测序目标区域
覆盖度(coverage)是指测序获得的序列占整个基因组的比例。样品来源不同,样品内的复杂度也不同,因此对不同样品会有不同的测序量要求。宏基因组项目交付合同说明的合理测序量,一般推荐简单环境(如哺乳动物肠道)测序数据量为 5G clean data;复杂环境(如土壤、海洋等)推荐数据量为 15G clean data。
3. 基础数据生成
过滤
无
组装
Q:TPM 能否用于不同样本之间的比较?
A:可以。根据 TPM 的原理,不同样本中所有基因 TPM 之和相等,因此 TPM 类似于物种注释结果中的相对丰度,是可以在不同样品/分组间进行比较的。
物种注释
Q:注释后物种丰度数据在哪里下载?
A:进入系统,点击页面顶部的下载分析结果按钮,您可以下载所有的分析结果,解压后您可以看到完整的物种丰度数据。
路径为:TaxonomyAnalysis/Rename_Abundance,其中:
- {分类水平}.absolute.xls 为 指定分类水平的绝对丰度表
- {分类水平}.relative.xls 为 指定分类水平的相对丰度表
- {分类水平}.{数据类型}.xls.header 为分析临时数据,不用管他
Q:为什么有的项目注释数量物种数目多,有的少?
A:物种注释结果数量大小与以下 3 点有关:
- 环境的复杂程度:环境越复杂,注释物种越多,反之亦然。土壤、水体等环境属于高复杂度环境,而肠道(粪便)样本属于低复杂度环境。
- 样本数量:随着样本数增加,样本中独有的物种被检测出来,会增加物种注释的总体数量。
- 测序深度:在测序数据量越高,越能把样品中稀有的物种注释出来。
若您测序结果中注释得到的物种很少,排除实验(实验污染)及测序(数据量)的问题,可能有以下原因:
- 所测的环境样品自身属于低复杂环境,含的物种较少
- 所送的样本组数太少,无法覆盖绝大部分物种
- 所关注的物种研究较少,数据库中物种的代表序列及相关信息较少
Q:注释结果中 NA 是什么意思?
A:宏基因组项目物种注释采用的是 Kraken2 软件,其原理是将测序序列打断成长度为 k(默认k=35)的子序列,称为 k-mer,然后将 k-mer 比对到自建数据库。比中数据库的 k-mer 会被分配一个到物种分类树上,没有比中的会被忽略,当一条序列的所有 k-mer 都有注释结果,再按照规则从注释树上选择最低层的物种注释结果即为这条序列的物种注释结果。但所有的 k-mer 可能只被注释到高层级,如属、科、纲,此时该序列的低层级会被标记为 NA。
Q:物种注释用数据库选择依据?
A:系统自动根据样本类型选择物种注释用数据库,若样本来源人肠道则选择 UHGG 数据,其他来源则选择 NCBI NT 数据库。
功能注释
Q:功能数据库的选择依据是什么?
A:Dr.Tom 系统提供了 KEGG、COG、eggNOG、Swiss-Prot、BacMet、CARD 和 CAZy 这 7 个常用数据库的功能注释结果,不同的数据库对于针对特定数据做了适当的优化,如 BacMet 关注杀菌剂和金属抗性基因,CARD关注抗生素抗性,您可以根据注释目的选择适当的物种注释结果。
Q:功能注释丰度表在哪里下载?
A:功能丰度表与 Clean Data 一起,线下交付。
Q:物种注释数据库版本?
A:不同功能数据库和数据的版本如下:
| 数据库 | 版本 |
|---|---|
| KEGG | v101.0 |
| COG | 20201125 |
| eggNOG | eggnog_5.0 |
| Swissprot | release-2021_04 |
| BacMet2 | 20180311 |
| Card | v3.0.9 |
| Cazy | 20240326 |
Q:数据库对菌群功能的注释是在属水平上进行的么?从发文章角度来说,我们只能自己选择其中一种数据库的结果来解释是么?
A:不是。功能注释不依赖于任何物种注释结果,功能注释依据序列的信息,这个你可以比对到多种库去进行分析,不一定只要一个库
Q:KEGG 注释结果中 map 和 pathway level3 是一一对应的,但丰度值却不一样?
A:map 和 pathway level3 采用了不同的统计算法:计算 map 时,若一个 KO 被注释到多个 map 则 KO 会被平均分配到各个 map 中;计算 pathway level3 时,直接累加 KO 丰度值。
4. 分析问题
物种/功能分布
Q:在统计物种/功能分布时,对数据做了什么处理?
A:通常,在一张图片显示太多的元素,会让这张图变得非常拥挤,我们很难从这样的图中得到有用的信息。基于此,需要对用于绘图的物种/功能丰度数据进行筛选:
- 归类到 others:符合筛选条件的数据直接保留,不符合条件的数据全部归类到 others 分类中。
- 物种/功能筛选:符合筛选条件的数据保留,不符合条件的直接舍弃。
筛选条件:
- 相对丰度前 N 的物种/功能
- 相对丰度大于 m 的物种/功能
请注意
物种/功能 归类到others 和 筛选 操作都是基于填写方案中的参数设定。但因为图形展示的原因,我们对部分图形最大展示物种/功能的数量做了限定。具体请查看对应页面说明。
物种累积曲线
Q:物种累积曲线具体怎么做的?有什么用?怎么看?
A:按照不同的分组,对组内的样本进行重采样,横坐标是重采样的数量,随着采样数量的增加,组内样本含有的数量逐渐增加,并进入一个平台期(增加曲线组间变缓),此时表明组内的样本数量足够多,多到可以代表这一分组的群落组成了,再增加组内的样本数量也不会增加组内物种的数量。
正是因为如此,所以物种累积曲线分析,对组内样本数量有要求。最少的样本数是 3,但仍然推荐数量越多越好。
多样性
Q:Bray-Curtis、JSD、欧氏距离有什么不同?如何选择?
A:欧式距离是对称指数,将双零数据视为相同的存在,另外两个是非对称指数,忽略双零数据。欧式距离的有一个特点是对数据的丰度数据的敏感性大于物种是否存在,并且欧式距离的计算方法决定了欧式距离的取值范围是无限大的。所以常用对于双零数据很多的物种丰度数据,选择 Bray-Curtis 和 JSD 会多一些,且 Bray-Curtis 相异性指数是最常用的指数。
差异分析
Q:Venn 图和 UpSetR 的选择依据?
A:根据分组的数量来进行选择的,当分组数小于 5 个时可以选择 Venn 图或者 UpSetR,当分组数大于 5 时 Venn 图不能展示那么多的数据了,只能选择 UpSetR 来绘图。
Q:参数检验和非参数检验有什么依据?
A:如果数据符合正态分布应该优先选用参数检验,其次才是非参数检验,因为非参数检验没有参数检验灵敏。如果数据中含有极端值,但因为一些原因无法去除,可以选用非参数检验,非参数检验对数据的排名进行检验,极端值对检验结果影响较小。
Q:组间显著性差异分析结果的解读 。
A:组间的差异分析是在不同的物种分类层级上组与组之间的差异是否显著的分析;通常采用的是假设检验, 验证其差异评价数据(例如平均数)相等的概率即 p 值,对 p 值的校正得到 q 值, 根据 p 值 或 q 值来判断组间的差异是否具有显著性。
Q:统计检验 p 值含义?
A:p 是统计检验 成立的概率。统计检验会提出两个互斥的假设, 称为零假设, 称为备择假设,而显著性检验的目的就是通过计算 p 值决定是接受 拒绝 ,还是接受 拒绝 。当 p < 0.05 时, 成立的概率已经很小了,所以接受 拒绝 。
举个例子,检验某个物种在不同分组中丰度是否差异。首先提出假设
- :该物种在两个分组中没有差异
- :该物种在两个分组中有差异
然后使用 Wilcoxon 检验或 T-test 计算 p 值,这个 p 值表示的是 成立的概率。若 p < 0.05,此时 成立的概率很低,而我们认为低概率事情不可能发生,所以就拒绝 ,认为该物种在两个分组中有差异。
小贴士
除非特别说明,Dr.Tom 系统默认的差异阈值 p 值为 0.05。
Q:为什么有的比较组中没有差异分析结果?
A:差异分析要求至少 2 个分组,每个分组中的生物学重复应该大于等于 3。对于不符合样本数量的比较组,不会做差异分析。
Q:想要更改 LEfSe 分组颜色
A:目前,系统没有提供 LEfSe 分组颜色更改功能。
Q:LEfSe 是怎么进行分析的?
A:LEfSe 分析有三个步骤:
- 首先利用 Kruskal-Wallis 秩和检验检测不同组间的物种丰度差异,获得差异显著特征(指定分类水平的分类);
- 对上一步的差异显著特征,对参与检验的分组进行两两比较,使用成组的 Wilcoxon秩和检验;
- 最后运用LDA(Linear discriminant analysis,线性判别分析)估计这些差异物种对组间区别的影响大小,即得到 LDA score。
Q:LEfSe 结果中 LDA 的含义?
A:LDA score 的含义是该物种对组间差异的贡献。
Q:LEfSe 方法筛选出来的主要是哪个水平的呢?
A:LEfSe 展示所有水平有差异物种。
Q:在富集分析中,两次自定义绘图或自定义绘图结果和已有结果之间,富集 Module 或 Pathway 和它对应的富集 p 值并不是完全一致,甚至富集结果中的数量都可能不一致,这是为什么?
A:这和富集分析的算法有关系,在富集分析时进行了置换检验,每次置换检验抽取的数据都是随机的,这会导致检验的结果有些许差异,而我们判断富集的标准固定的,因此富集到的 Module/Pathway 数量可能会发生变化。因此建议您关注那些差异比较明显的结果。
物种关联
Q:怎样选择用 CCA 还是 RDA 来进行环境因子对群落结构影响的展示?
A:RDA 或 CCA 模型的选择原则:RDA 是基于线性模型,CCA 是基于单峰模型。一般会选择 CCA 来做直接梯度分析。但是如果 CCA 排序的效果不太好,就可以考虑用 RDA 分析。
先用物种丰度数据做 DCA(Detrended Correspondence Analysis) 分析,分析结果中 Lengths of Gradient 的第一轴的大小。如果大于 4.0,就应该选 CCA;如果 3.0-4.0 之间,选 RDA 和 CCA 均可;如果小于 3.0,RDA 的结果要好于 CCA。
Q:CCA 和 RDA 环境因子数必须小于样本数的原因是什么?
A:二者都属于约束性排序, CCA 属于非线性模型;RDA 属于线性模型;先看 PCA 的排序, 其未设置环境因子是一种非限制性, 所找到的主成分是可以说一种未知的环境变量; 而约束性排序是指在特定的环境变量(环境因子)上进行排序,即提供坐标轴方向进行排序; 如有n个样品,提供n个坐标方向,就起不到这种特定方向的约束的作用了。
参考文献:
Legendre, P. and L. Legendre (2012). Numerical Ecology, 3rd English ed. Amsterdam: Elsevier Science BV.
Q:CCA 和 RDA 的变量要求是具体数值的原因是什么?
A:CCA 和 RDA 是环境因子的关联性分析,算法模型要求环境因子数据是连续性的(数据是有大小、顺序的数字),比如测定的 pH 值,温度等。 同时非连续型变量不能作为环境因子数据;比如说:对比用药组和对照组的不同,这是将加入了药物记为 1, 没有加入的记为 0,这类变量属于非连续性变量 (或者叫离散型),可以作为分组变量。
Q:Pearson 相关系数和 Spearman 相关系数的区别?
A:Pearson 相关系数适用于定量数据,要求数据满足正态分布、数据之间有线性相关,Spearman 相关系数适用于定量或等级数据,数据符合任何分布模式均可,参与计算的数据通常为非线性相关。
小贴士
除非特别说明,Dr.Tom 默认使用的是 Spearnman 相关性系数,阈值为相关性系数绝对值大于 0.3 且 p 值小于 0.05
其他分析
Q:什么是置换检验?
A:通过进行实验和采样,获得了研究对象的部分数据,然后用这个数据来描述我们需要研究对象的整体。我们知道采样的样本量越大,对整体的描述就越准确,但实际实验经过程中,因为各种原因常得到的样本数量有限。当样本量很小,分布未知,如何用来描述总体呢,这个时候就需要用到置换检验。
置换检验(Permutation test)本质上是重抽样方法,它将全部(或部分)样本数据随机抽样排列,然后将抽样获得的样本统计量和实际观测到的样本统计量进行比较,通过大量的置换(R 中默认 999 次),计算置换后的统计量大于实际观测到的统计量的概率,即为置换检验 p 值,根据 p 值进行统计推断。这个方法最初由 Fisher 在 20 世纪 30 年代提出。
提示
因为置换检验进行的是随机抽样,所以多次置换检验的结果并不完全一致。