小工具
关联转换
调出与选定的数据有对应关系(互作关系、映射关系、位置关系等)的数据,以表格形式展现。
互作关系-PPI
介绍: 预测蛋白质与蛋白质之间的互作关系。调出与所选基因/转录本/蛋白有互作关系的基因/转录本/蛋白。
数据库: 基于 STRING11 已知数据库结果和 NCBI Reference 的转录本映射关系得到结果; 分别从 STRING 数据库 (https://string-db.org/cgi/download.pl?sessionId=esF2YhHLpzd0) 和 NCBI Reference (https://www.ncbi.nlm.nih.gov/) 获得对应版本的数据。
参数解释:
- min score:基于 STRING11 数据库的打分,分值为 0-1000。打分越高,得到的 PPI 关系越可能准确,关联出的 mRNA 越少。默认设置为 500。
- Limit:转换得到的基因 / 转录本 / 蛋白数目有限制。通过 max num 限制得到的基因 / 转录本 / 蛋白的最大数目。
- No limit:转换得到的基因 / 转录本 / 蛋白数目没有限制。
结果展示:
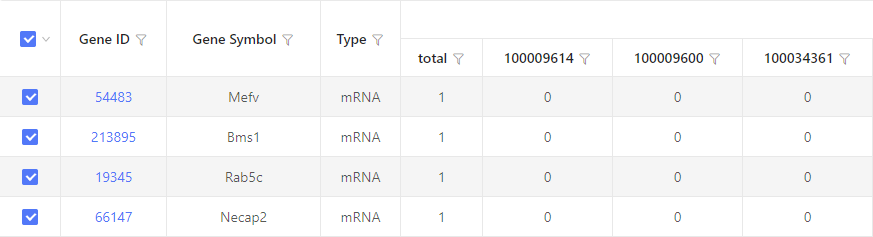
以表格形式展示,行为所选基因 / 转录本 / 蛋白,列为与所选基因 / 蛋白 / 转录本有互作关系的基因。数字 “1“表示行列有互作关系,数字 “0” 表示行列无互作关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
互作关系 - Target
介绍: 预测与 microRNA 有靶向关系的 mRNA/lncRNA/circRNA。其中,circRNA 仅限人、小鼠。
预测软件: circRNA 与 miRNA 的靶向关系直接从 starbase 数据库获取;lncRNA, mRNA 与 miRNA 的靶向关系预测使用的是同一套预测软件;
动物: TargetScan:默认参数 RNAhybrid 和 miRanda 可用做所有动物的靶向关系预测。其中 - s 可选为 3utrfly,3utrworm,3utr_human miRanda:-en -20 -strict RNAhybrid:=-b 100 -c -f 2,8 -m 100000 -v 3 -u 3 -e -20 -p 1 -s 3utr_human
植物 / 细菌 / 真菌: tapir: --score 5 --mfe_ratio 0.6 targetfinder: -c 4
参数解释:
- min score:基于预测到的软件个数,给该预测结果打分。对于人或动物,由于预测到的靶基因较多,只保留了≥2 个软件共同预测到的靶基因,即 score=2 或 3;对于植物 / 细菌 / 微生物,score=1 或 2。Min score=n,即表明仅筛选有 ≥n 个软件共同预测到的靶基因。另外,由于 circRNA 与 miRNA 靶向关系为直接从公共数据库中获取,其分值默认 = 3。
- Limit:转换得到的基因数目有限制。通过 max num 限制得到的基因的最大数目。
- No limit:转换得到的基因数目没有限制。
结果展示:
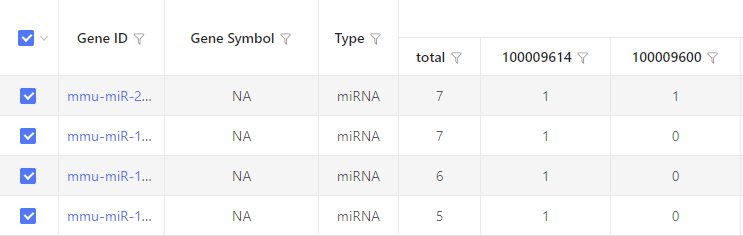
以表格形式展示。“total” 表示有靶向关系的基因总数。数字 “1” 表示有靶向关系,数字 “0” 表示无靶向关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
互作关系 - rnaplex
介绍: 通过 rnaplex 软件预测 RNA 之间的相互作用。
参考:Tafer H, Hofacker IL. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics. 2008 Nov 15;24(22):2657-63. doi: 10.1093/bioinformatics/btn193.
参数解释:
- max MFE:rnaplex 软预测中使用的最大自由能,数值范围为无穷小到 - 100。值越小,结合越紧密,得到的关系可能越准确,得到的 lncRNA/mRNA 更少。
- Limit:转换得到的基因数目有限制。通过 max num 限制得到的基因的最大数目。
- No limit:转换得到的基因数目没有限制。
结果展示:
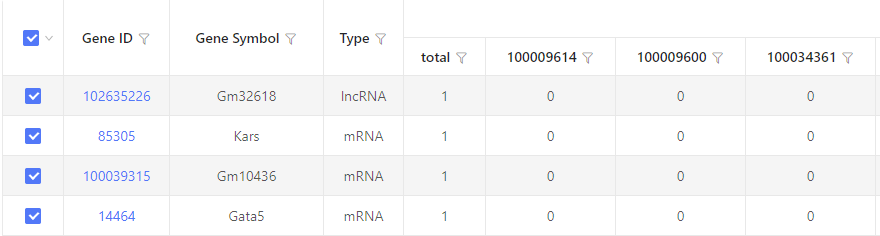 以表格形式展示。“total” 表示有互作关系的基因总数。数字 “1“表示有互作关系,数字 “0” 表示无互作关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
以表格形式展示。“total” 表示有互作关系的基因总数。数字 “1“表示有互作关系,数字 “0” 表示无互作关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
互作关系-GGI
介绍: 预测有关联的基因(仅针对人、小鼠)。
软件原理:用 NCBI Pubmed 数据库的文献摘要,用机器学习相关算法进行文本挖掘,识别出有关联的基因,并作出标记。根据人测试数据集的统计,precession 和 recall 均在 80% 以上。
参数解释:
- min score:最小关联度分值。分值设置范围为 1-586,分值越大,关联度越强。
- Limit:转换得到的基因数目有限制。通过 max num 限制得到的基因的最大数目。
- No limit:转换得到的基因数目没有限制。
结果展示:
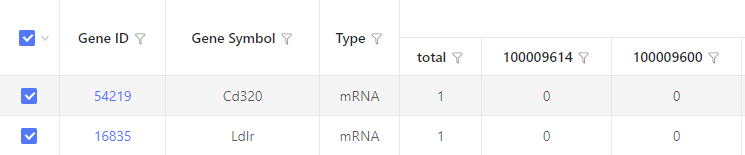 以表格形式展示。“total” 表示与该行对应的基因有互作关系的基因总数。数字 “1“表示有互作关系,数字 “0” 表示无互作关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
以表格形式展示。“total” 表示与该行对应的基因有互作关系的基因总数。数字 “1“表示有互作关系,数字 “0” 表示无互作关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
互作关系-ceRNA
介绍: 调出选定 mRNA/LncRNA 的 ceRNA。暂时只支持人、小鼠。
原理:
- 根据靶向关系得到 RNAx 和 RNAy 的矩阵,根据共享 miRNA 数量,RNAx, RNAy 的靶向 miRNA 数量和所有 miRNA 的数量计算显著性 P 值和 Q 值 (RNAx,RNAy 分别指一对关系中靶向和被靶向的 RNA)
- 计算统计了 RNAy 与 RNAx 的共享 miRNA 比例 (共享 miRNA 数量 / RNAx 的靶向 miRNA 数量) 并保留下 top10% 的关系对
- 用结果关系对和其对应的 Pvalue, Qvalue 作为最后结果,并根据 Qvalue 计算 score
Q value=10-2~1,score = -log(Qvalue)*44
Q value=10-12~10-2,score =88 + (-log(Qvalue))
Q value=0~10-12,score=100
参数解释:
- min score:最小得分值,范围为0-100。分值越高,ceRNA关系越可靠。
- Limit:转换得到的基因数目有限制。通过max num限制得到的基因的最大数目。
- No limit:转换得到的基因数目没有限制。
结果展示:
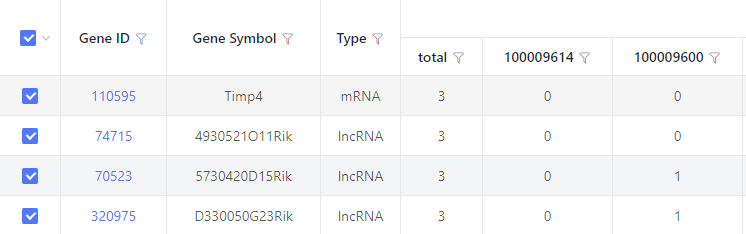 以表格形式展示。“total” 表示与该 RNA 互为 ceRNA 的 RNA 总数。数字 “1“表示行列互为 ceRNA,数字 “0” 表示行列无 ceRNA 关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
以表格形式展示。“total” 表示与该 RNA 互为 ceRNA 的 RNA 总数。数字 “1“表示行列互为 ceRNA,数字 “0” 表示行列无 ceRNA 关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
映射关系-gene/transcript to protein
介绍: 调出基因/转录本对应的蛋白。
结果展示:
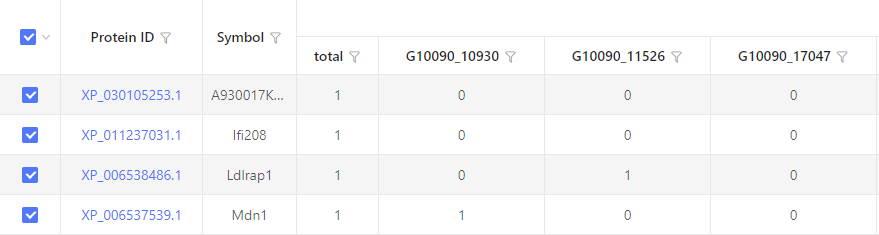 以表格形式展示。横行为选中的基因 / 转录本,纵列为蛋白。“total” 表示与基因 / 转录本对应的蛋白总数。数字 “1“表示该行基因 / 转录本与该列蛋白相互对应,数字 “0” 表示行列无对应关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
以表格形式展示。横行为选中的基因 / 转录本,纵列为蛋白。“total” 表示与基因 / 转录本对应的蛋白总数。数字 “1“表示该行基因 / 转录本与该列蛋白相互对应,数字 “0” 表示行列无对应关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
映射关系-cell to gene
介绍: 调出选定细胞中检测到的基因。
参数解释:
表达量:设置表达量阈值,挑选出表达量高于该设定阈值的基因。
结果展示:
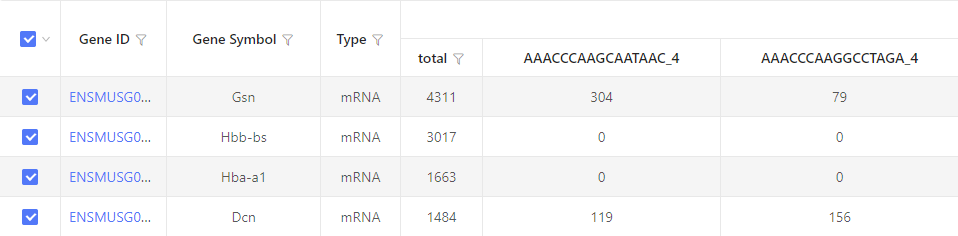 以表格形式展示。纵列为选中的细胞,横行为检测到的基因。“total” 表示该基因在所有细胞中检测到的 UMI 总数。数字表示该细胞中检测到对应基因的 UMI 数目。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
以表格形式展示。纵列为选中的细胞,横行为检测到的基因。“total” 表示该基因在所有细胞中检测到的 UMI 总数。数字表示该细胞中检测到对应基因的 UMI 数目。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
映射关系-gene to cell
介绍: 调出检测到选定基因的细胞。
参数解释:
- 选择样本:只可选择单一样本。
- 表达量:设置表达量阈值,挑选出选定基因表达量高于该设定阈值的细胞。
结果展示:
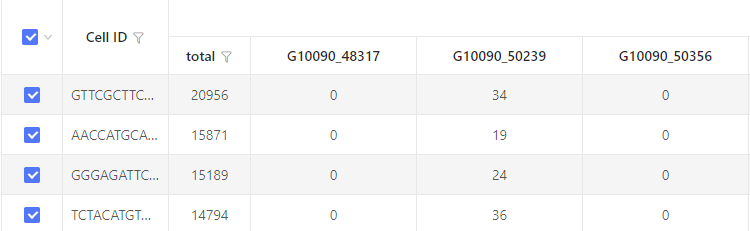
以表格形式展示。纵列为选中的基因,横行为对应的细胞。“total” 表示该细胞中检测到的所有基因的 UMI 总数。数字表示该细胞中检测到对应基因的 UMI 数目。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
位置关系
介绍: 调出与选定的基因/转录本有位置关系的基因/转录本。可调整上下游范围参数及正、反链。结果以表格形式展现。
位置关系-DMR to gene/DMR to transcript
介绍: 调出与选定的差异甲基化区域(DMR)有位置关系的基因 / 转录本。可调整上下游范围参数。
结果展示:
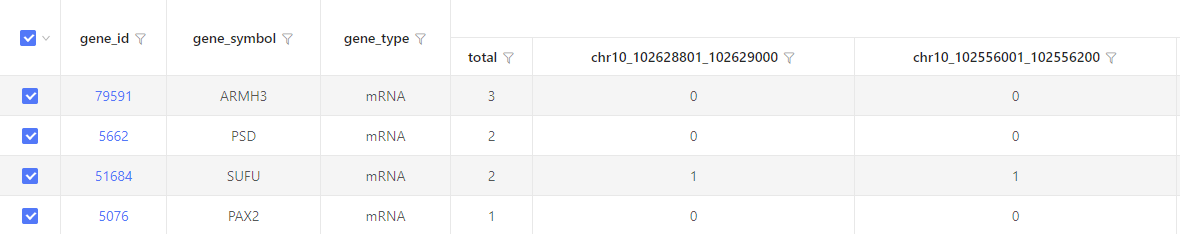 以表格形式展示。纵列为选中的 DMR,横行表示基因 / 转录本。“total” 表示对应到该基因 / 转录本的 DMR 总数。数字 “1“表示该行基因 / 转录本与该列 DMR 相互对应,数字 “0” 表示行列无对应关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
以表格形式展示。纵列为选中的 DMR,横行表示基因 / 转录本。“total” 表示对应到该基因 / 转录本的 DMR 总数。数字 “1“表示该行基因 / 转录本与该列 DMR 相互对应,数字 “0” 表示行列无对应关系。默认显示 20 列,超出部分将在界面中隐藏。通过下载表格可查看隐藏部分。
富集
工具介绍:进行 KEGG、GO 等富集分析,帮助了解所选定基因在哪些代谢通路、生物学过程中发生富集。
原理:
富集分析采用超几何检验,找出与全部基因背景相比,在候选基因中显著性富集的通路/功能等,计算公式为:
N为所有基因中,在所选数据库中具有注释的基因数目; n为N中候选基因的数目; M为所有基因中注释为某特定通路/功能的基因数目; m为注释为某特定通路/功能的候选基因数目。
通常 Qvalue ≤ 0.05的通路/功能视为显著富集
软件:R 软件中的 phyper 函数。
数据库介绍:
KEGG Pathway:是有关 Pathway 的主要公共数据库,基于对生命活动中的分子相互作用和化学物质的反应的认识,构建了复杂的调控网络,绘制成通路。通过 Pathway 显著性富集能确定候选基因参与的最主要生化代谢途径和信号转导途径。
GO:Gene Ontology,是一个国际标准化的基因功能分类体系,提供了一套动态更新的标准词汇表(controlled vocabulary)来全面描述生物体中基因和基因产物的属性。GO 总共有三个 ontology(本体),分别描述基因的分子功能(molecular function)、细胞成分(cellular component)、参与的生物过程(biological process)。GO 的基本单位是 term(词条、节点),每个 term 都对应一个属性。通过 GO 功能显著性富集分析能确定候选基因行使的主要生物学功能。
MSigDB:分子标签数据库,共包含 8 个系列。仅针对人物种提供结果。
- H: hallmark gene sets(癌症)特征基因集合,共 50 组;
- C1: positional gene sets 位置基因集合,根据染色体位置,共 326 个;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等;
- C3: motif gene sets:模式基因集合,主要包括 microRNA 和转录因子靶基因两部分;
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括 BP(生物学过程 biological process,细胞原件 cellular component 和分子功能 molecular function 三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于 NCBI GEO 发表的芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
Transcription Factor:植物的已知转录因子信息来自 PlantTFDB v5.0;动物的已知转录因子来自 AnimalTFDB v3.0。动物除了 TF 信息外,还有 TF Cofactors 信息,同样来自 AnimalTFDB v3.0。
AnimalTFDB v3.0: 包含 97 个物种全基因组水平的 125,135 个 TF 基因和 80,060 个转录辅因子基因。其中,根据转录因子 DNA 结合结构域(DBD),将 TF 进一步分为 73 个家族,根据 TF 辅助因子功能分为 83 个家族。
PlantTFDB v5.0:包含了从 165 个物种中预测的 320,370 个 TFs,并将其分成 58 个家族。
Pfam:Pfam(Protein families database of alignments and hidden Markov models )提供了完整准确的蛋白质家族和结构域分类信息,以多序列比对信息和隐马尔可夫模型(HMM)表示,广泛应用于蛋白家族查询和蛋白结构域的注释。
Reactome:Reactome 是开源的关系数据库,它记录了信号和代谢分子及其组成生物途径和过程的关系。Reactome 数据模型的核心单元是反应 (reaction)。参与反应的物质 (核酸、蛋白质、复合物、小分子等) 形成了生物相互作用的网络,并被归类为途径 (pathway)。Reactome 中的生物途径包括代谢、信号转导、转录调控、凋亡和疾病。Reactome 的通路都是由专家撰写,经同行评阅的有关人体内各项反应及生物学路径的文章的数据库,目前在医学、遗传学、基因组学、分子生物学等领域的研究人员用于解释高通量实验研究的结果。
COG:COG 数据库是 NCBI 开发的用于同源蛋白注释的数据库,是将细菌、藻类和真核生物的 21 个完整基因组的编码蛋白,根据系统进化关系分类构建而成。通过鉴定蛋白与数据库的比对,可以很好的预测蛋白质的功能。
EggNOG:直系同源蛋白分组比对(Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups)数据库,由 EMBL 创建维护,是对 NCBI 的 COG 数据库进行拓展,提供不同分类水平蛋白的直系同源分组(Orthologous Groups,OG),包括真核、原核及病毒的数据信息。它扩展了 COG 数据库的分类方法,采用无监督聚类算法在全基因组范围内推导基因功能,更适用于谱系特征基因的分析。
InterPro:InterPro 是集成的蛋白质结构域和功能位点数据库,包含关于蛋白质家族、域、重复序列、和作用位点等数据资源,同时,InterPro 也包含很多来自不同数据库的诊断签名的人工注释文件,形成了一个给定的蛋白质家族、结构域和功能位点的独特描述。
使用示例
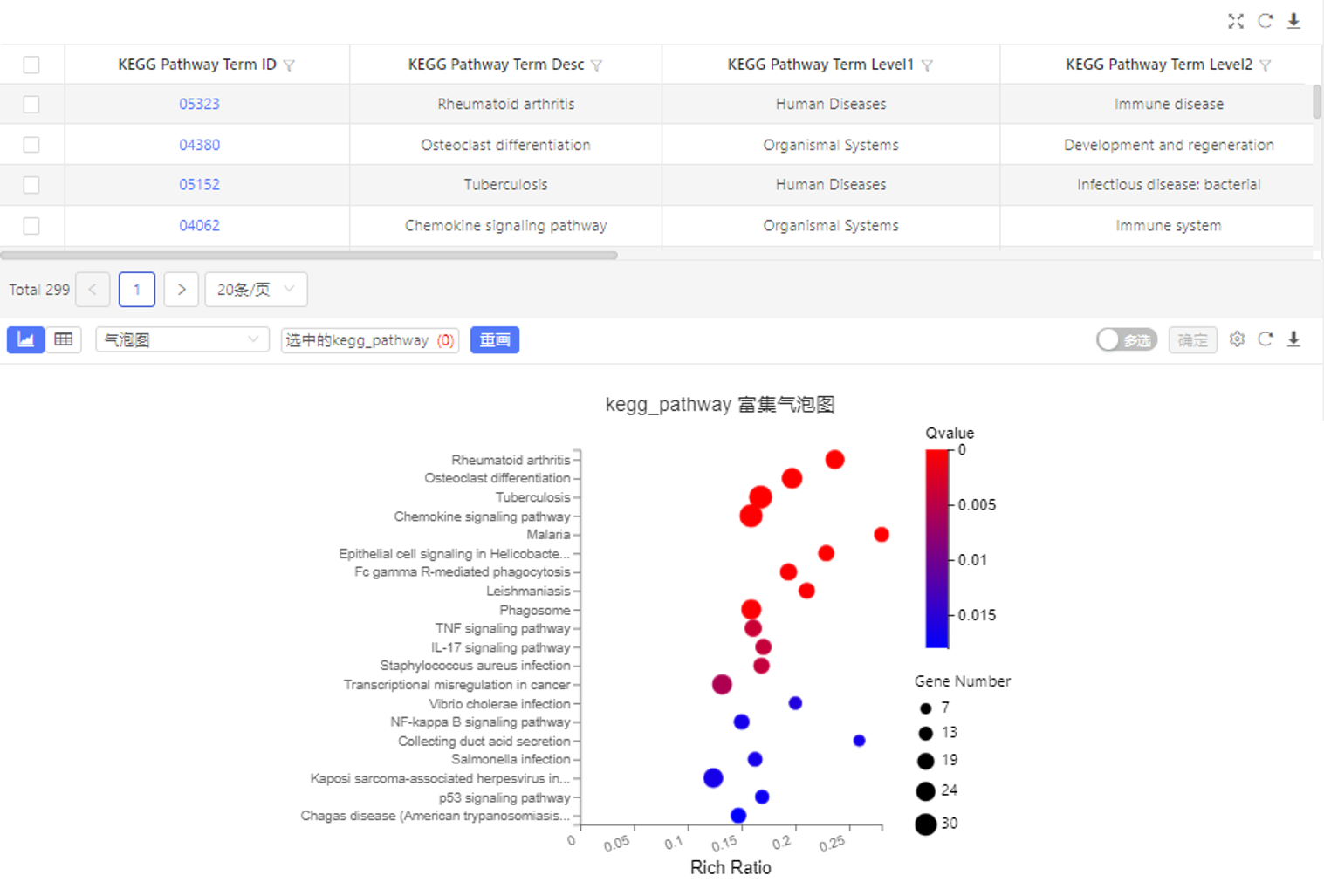 表格中列出所选定的基因富集到的通路,图片默认展示 q-value 从小到大排序前 20 条的通路。
表格中列出所选定的基因富集到的通路,图片默认展示 q-value 从小到大排序前 20 条的通路。
GSEA富集
工具介绍:使用 Broad Institute 开发的基因集富集分析(GSEA)算法进行分析,帮助识别所选择的样本中表达量变化最明显的基因集中在哪些通路、生物学过程或功能组分等。
原理: 使用预定义的基因集(系统提供 GSEA MSigDB、KEGG、GO、Pfam、Reactome、Transcript Factor、TF Cofactors、Cog、EggNOG),将基因按照在两类样本中的差异表达程度排序,然后检验预先设定的基因集合是否在这个排序表的顶端或者底端富集。
原理详见 https://www.gsea-msigdb.org/gsea/doc/subramanian_tamayo_gsea_pnas.pdf
软件:GSEA官方软件包https://www.gsea-msigdb.org/gsea/index.jsp
参数解释:
过滤阈值
- Max size (exclude larger sets):通路中包含基因数的最大值
- Min size (exclude smaller sets):通路中包含基因数的最小值
使用示例
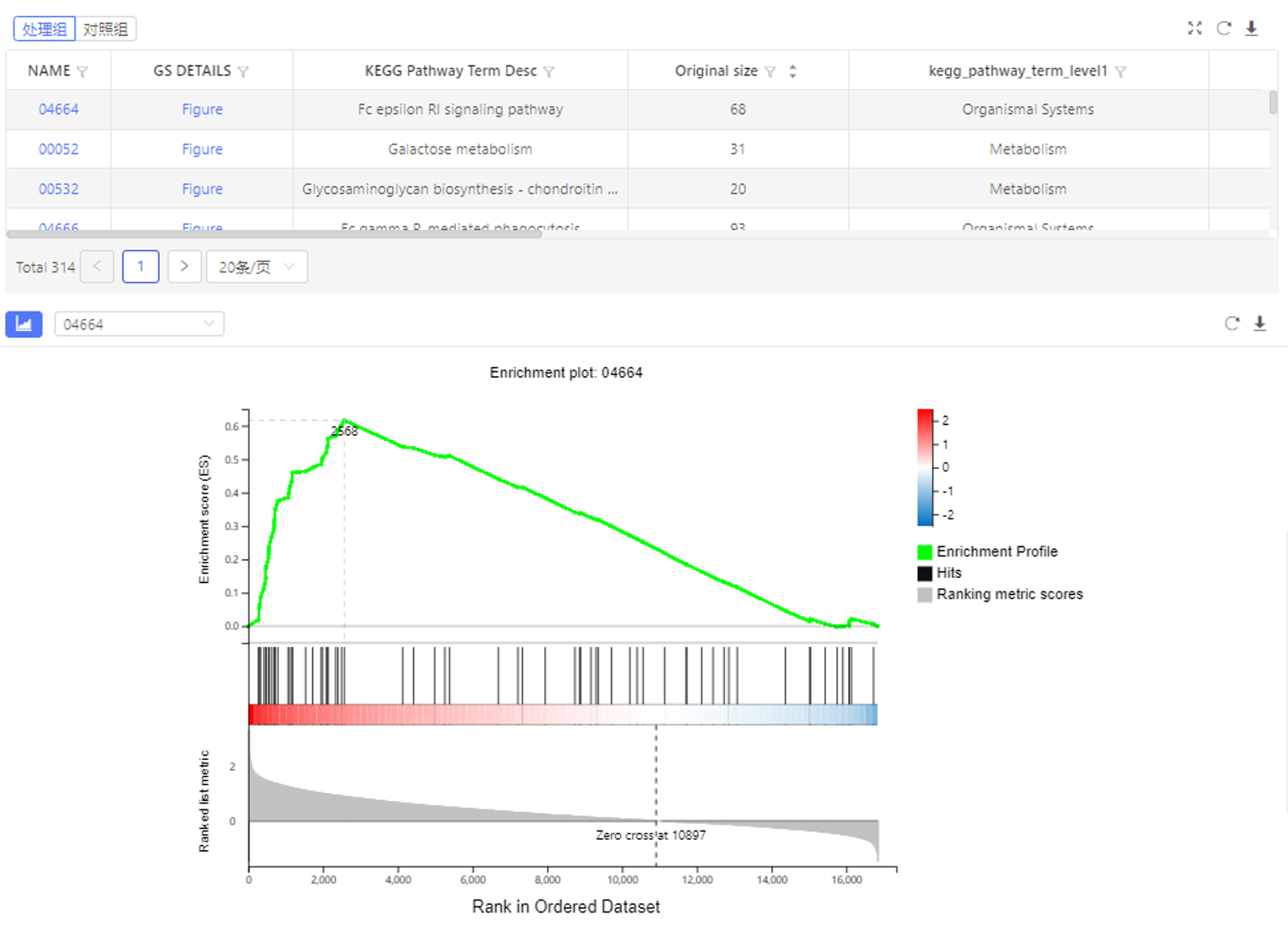 表格中列出预定义基因集在本次富集中富集到的基因个数、富集得分、名义 P 值等相关信息。图片展示 GSEA 富集结果(将标准化的富集得分从高到低排序,系统仅展示标准化富集得分排名前 100 的基因集的 GSEA 富集结果图)。
表格中列出预定义基因集在本次富集中富集到的基因个数、富集得分、名义 P 值等相关信息。图片展示 GSEA 富集结果(将标准化的富集得分从高到低排序,系统仅展示标准化富集得分排名前 100 的基因集的 GSEA 富集结果图)。
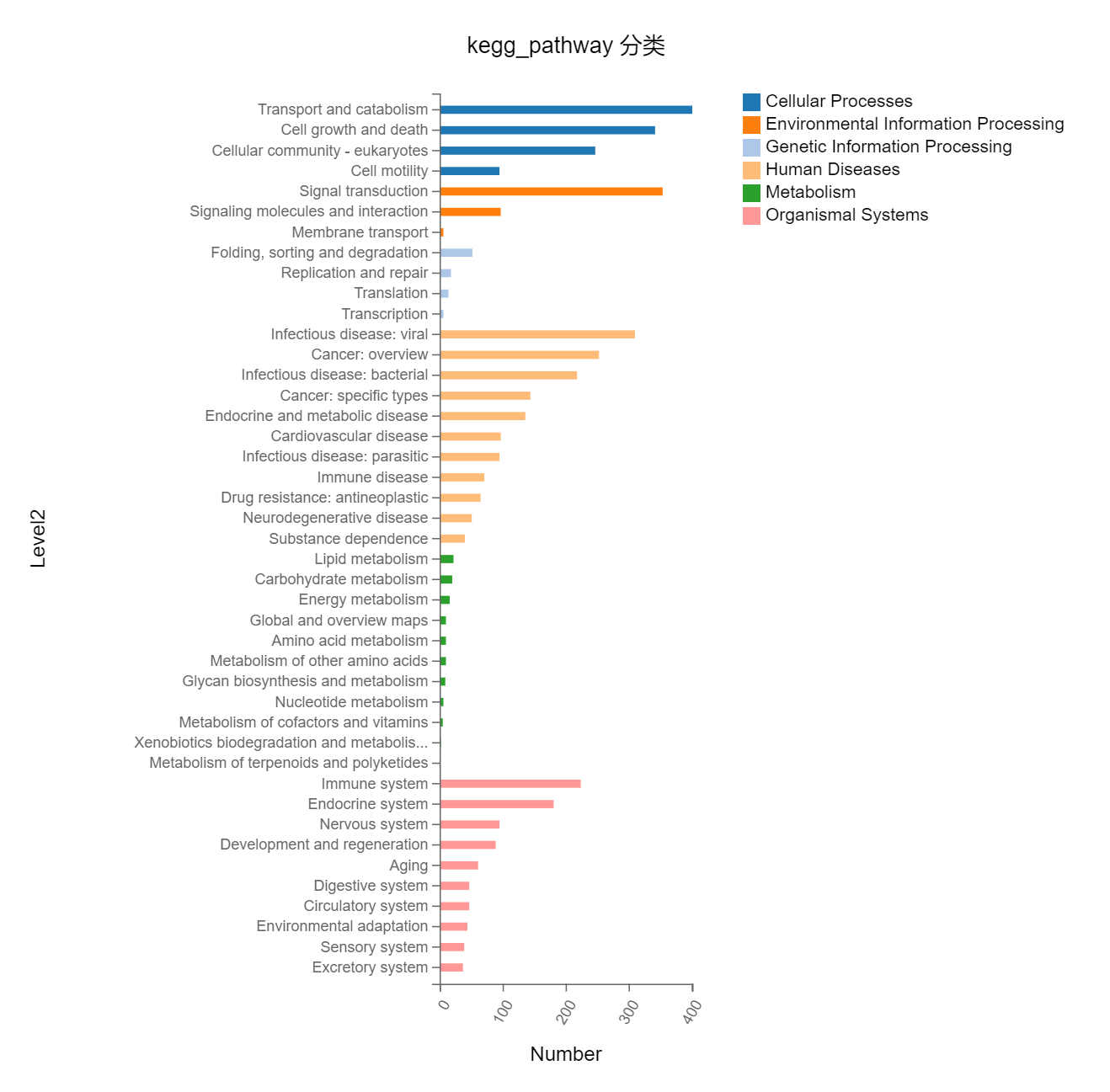
分类
工具介绍:进行注释分类(系统提供 GSEA MSigDB、KEGG、GO、Pfam、Reactome、Transcript Factor、TF Cofactors、Cog、EggNOG 等数据库注释),绘制条形图。查看选定基因在数据库中注释到哪些条目。
使用示例
聚类热图
工具介绍:使用表达量、差异倍数或自定义数据绘制聚类热图,帮助了解多样本、多个基因的表达量变化及聚类关系。
使用示例
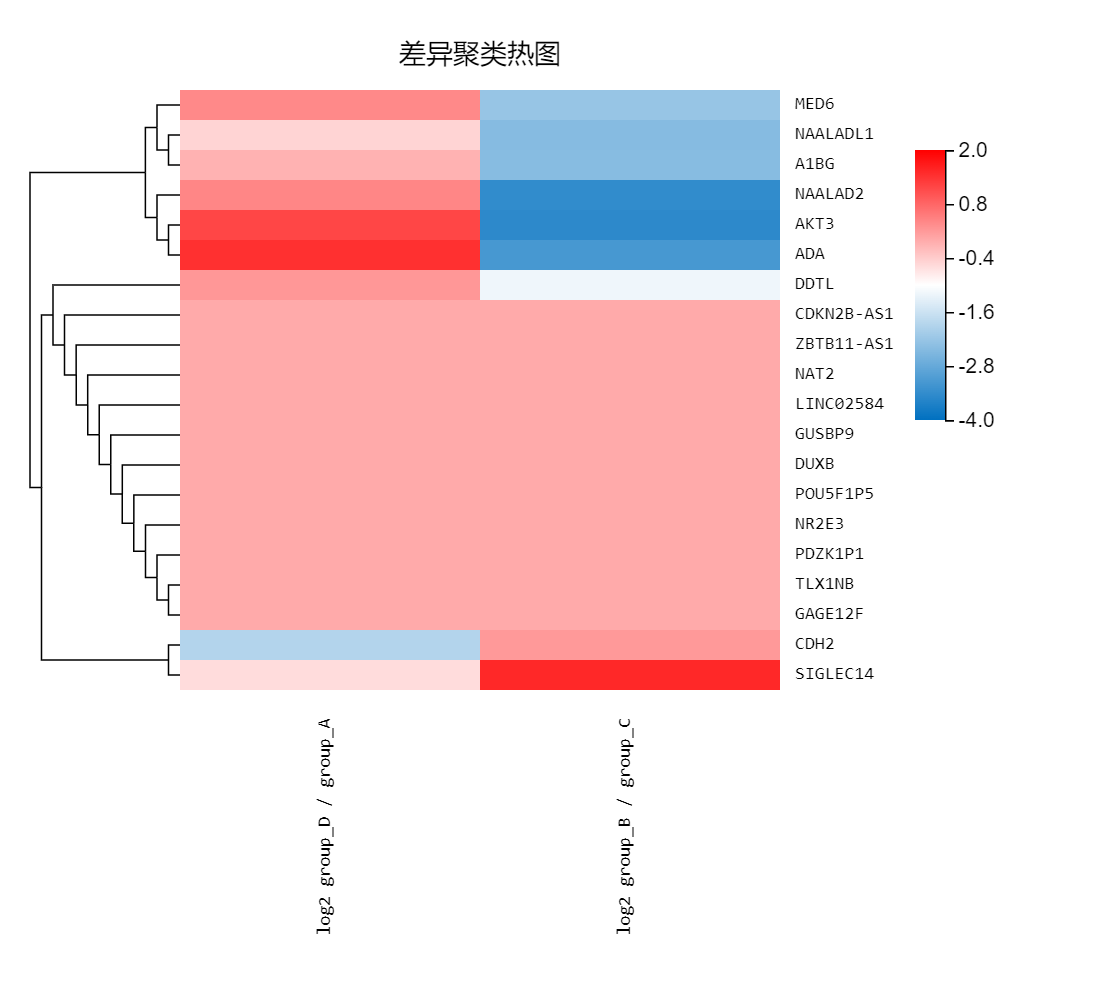 展示聚类热图。可通过图片设置增加行标签及列标签,或增加第三维度信息进行作图。所有图片可调整大小、颜色等,标题文字可修改。
展示聚类热图。可通过图片设置增加行标签及列标签,或增加第三维度信息进行作图。所有图片可调整大小、颜色等,标题文字可修改。
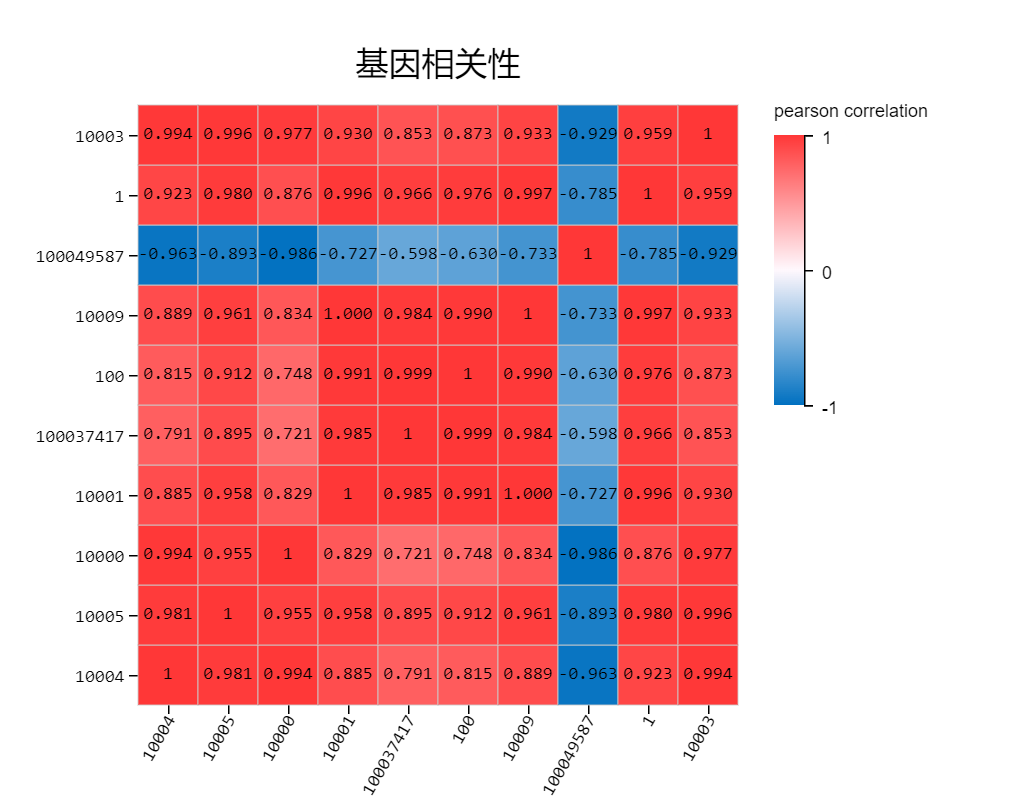
基因相关性
工具介绍:根据所选基因的 TPM/FPKM/Read Count 值,选择 Pearson 或 Spearman 相关性系数算法,绘制基因相关性热图,帮助了解基因间的相关性。
原理介绍:
Pearson 相关系数:皮尔森相关系数(Pearson correlation coefficient)也称皮尔森积差相关系数 (Pearson product-moment correlation coefficient) ,是一种线性相关系数。皮尔森相关系数是用来反映两个变量线性相关程度的统计量。相关系数用 r 表示,r 描述的是两个变量间线性相关强弱的程度。r 的绝对值越大表明相关性越强。
n 为样本量, X,Y 分别为两个变量的观测值和均值。
Spearman 相关系数:斯皮尔曼等级相关(Spearman’s correlation coefficient for ranked data),无参数的等级相关系数,亦即其值与两个相关变量的具体值无关,而仅仅与其值之间的大小关系有关。di 表示两个变量分别排序后成对的变量位置差,N 表示 N 个样本,减少异常值的影响。
使用示例
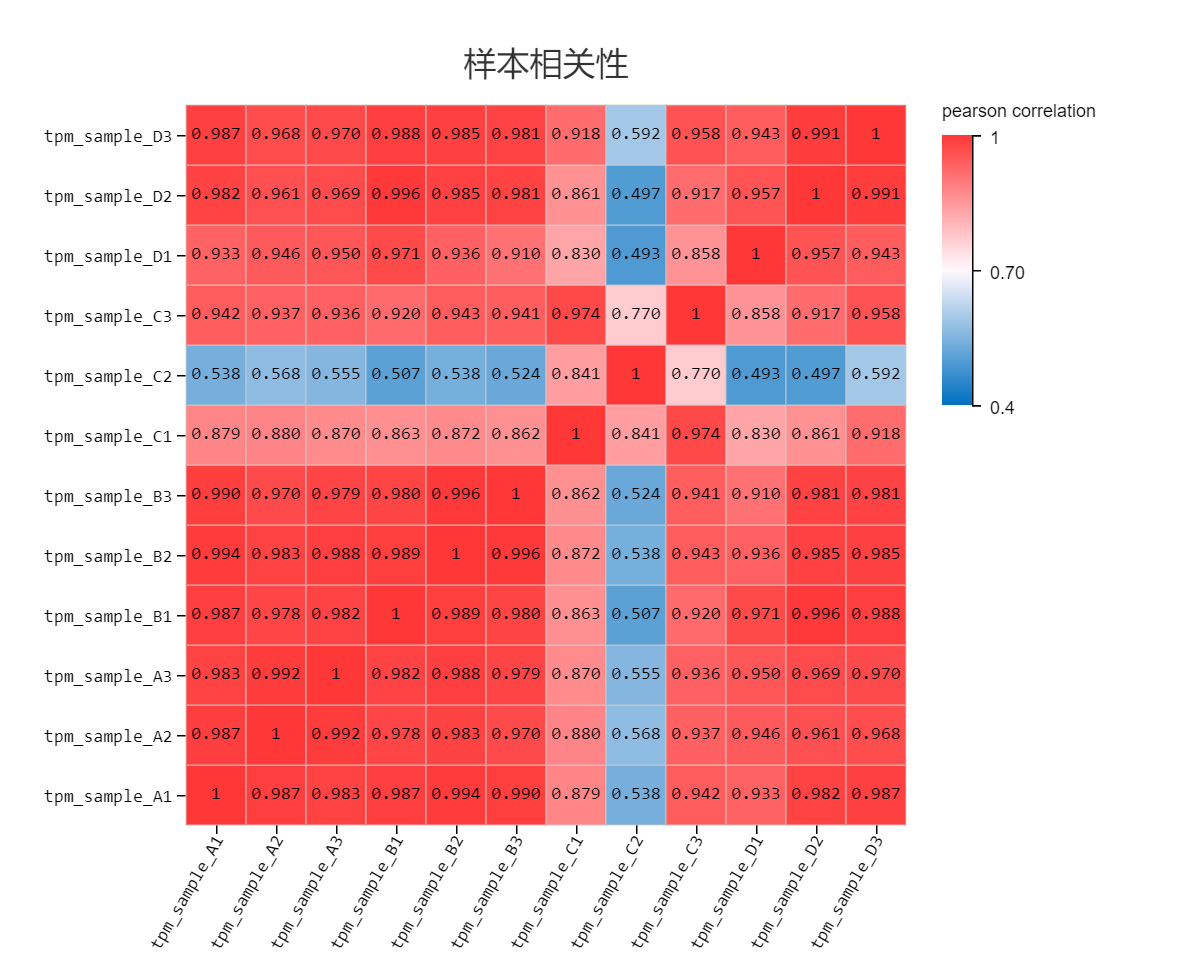
样本相关性
工具介绍:根据选定基因的 TPM/FPKM/Read Count 值,选择 Pearson 或 Spearman 相关性系数算法,绘制样本相关性热图,帮助了解样本间的相关性。
原理介绍:
Pearson 相关系数 皮尔森相关系数(Pearson correlation coefficient)也称皮尔森积差相关系数 (Pearson product-moment correlation coefficient) ,是一种线性相关系数。皮尔森相关系数是用来反映两个变量线性相关程度的统计量。相关系数用 r 表示,r 描述的是两个变量间线性相关强弱的程度。r 的绝对值越大表明相关性越强。
n 为样本量 X,Y 分别为两个变量的观测值和均值
Spearman 相关系数 斯皮尔曼等级相关(Spearman’s correlation coefficient for ranked data),无参数的等级相关系数,亦即其值与两个相关变量的具体值无关,而仅仅与其值之间的大小关系有关。di 表示两个变量分别排序后成对的变量位置差,N 表示 N 个样本,减少异常值的影响。
使用示例
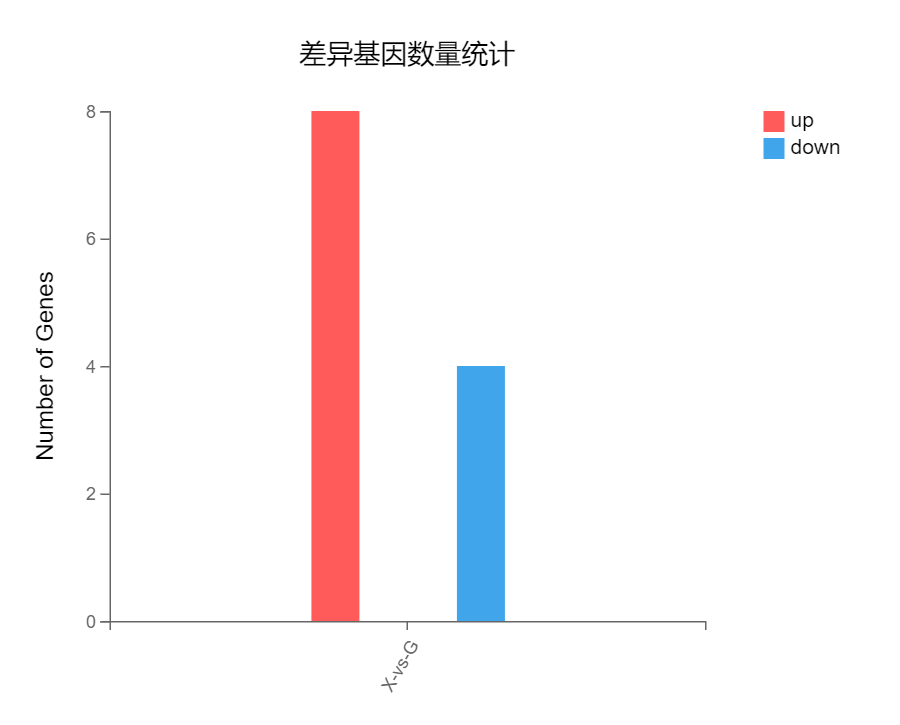
差异分析
工具介绍:对自定义的两个组或两个样本做差异基因分析,分析结果以条形图展示。支持有生物学重复(DESeq2/DEGseq/edgeR)/ 无生物学重复(DEGseq/PossionDis)样本的分析。
软件及适用范围:
DESeq2 仅支持有生物学重复的样本进行差异分析。 Love, M.I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). https://doi.org/10.1186/s13059-014-0550-8
DEGseq 支持有生物学重复/无生物学重复的样本进行差异分析。 Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010 Jan 1;26(1):136-8. doi: 10.1093/bioinformatics/btp612.
edgeR 仅支持有生物学重复的样本进行差异分析。 Mark D. Robinson, Davis J. McCarthy, Gordon K. Smyth, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, Volume 26, Issue 1, 1 January 2010, Pages 139–140, https://doi.org/10.1093/bioinformatics/btp616
PossionDis 仅支持无生物学重复的样本进行差异分析。 Audic, S. and J. M. Claverie. (1997). The significance of digital gene expression profiles. Genome Res, 10: 986-95.
使用示例
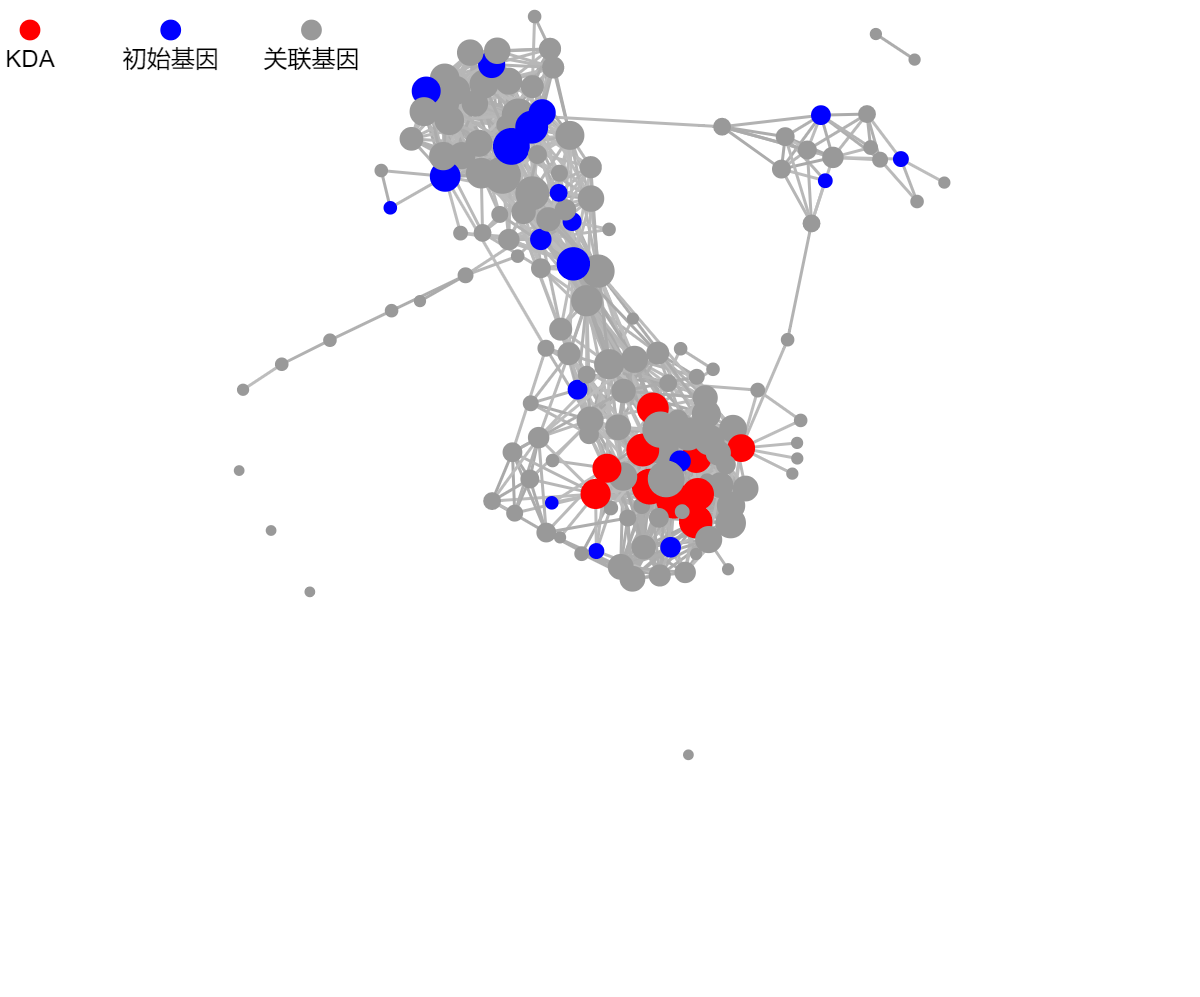
网络互作
工具介绍:绘制网络互作图,帮助了解选定的基因间的互作、共表达、调控等关系。支持 PPI、microRNA 靶向、ceRNA、rnaplex 及 GGI 互作关系的单独或同时展示。
参数解释 : (详见对应 PPI\Targe\ceRNA\rnaplex\GGI 参数解释)
使用示例
图片展示选定的基因之间的关系。不同的连线颜色表示不同的互作关系,不同的图形代表不同的 RNA 类型。图形颜色和大小默认代表连接数,可通过图设置进行调整。

KDA
工具介绍:进行关键驱动基因分析,帮助了解所选择的基因中处于调控主要地位的基因。
参数解释 :
- 关键基因:拖动滑杆调节关键基因数目
- 关联基因:拖动滑杆调节关联基因数目
- Score:基于 STRING11 数据库的打分,分值为 400-1000(400 分以下的不会计入结果)。打分越高,得到的关系越可能准确。默认设置为 500。
使用示例
KEGG网络图
工具介绍:绘制 KEGG 网络互作图,以网络图的形式展示所选基因与 KEGG Pathway 关系。
使用示例
 正方形表示 KEGG 通路,其他形状表示不同的 RNA 类型,图形颜色和大小默认代表连接数,可通过图设置进行调整。连线颜色表示不同的 KEGG 通路类别。
正方形表示 KEGG 通路,其他形状表示不同的 RNA 类型,图形颜色和大小默认代表连接数,可通过图设置进行调整。连线颜色表示不同的 KEGG 通路类别。
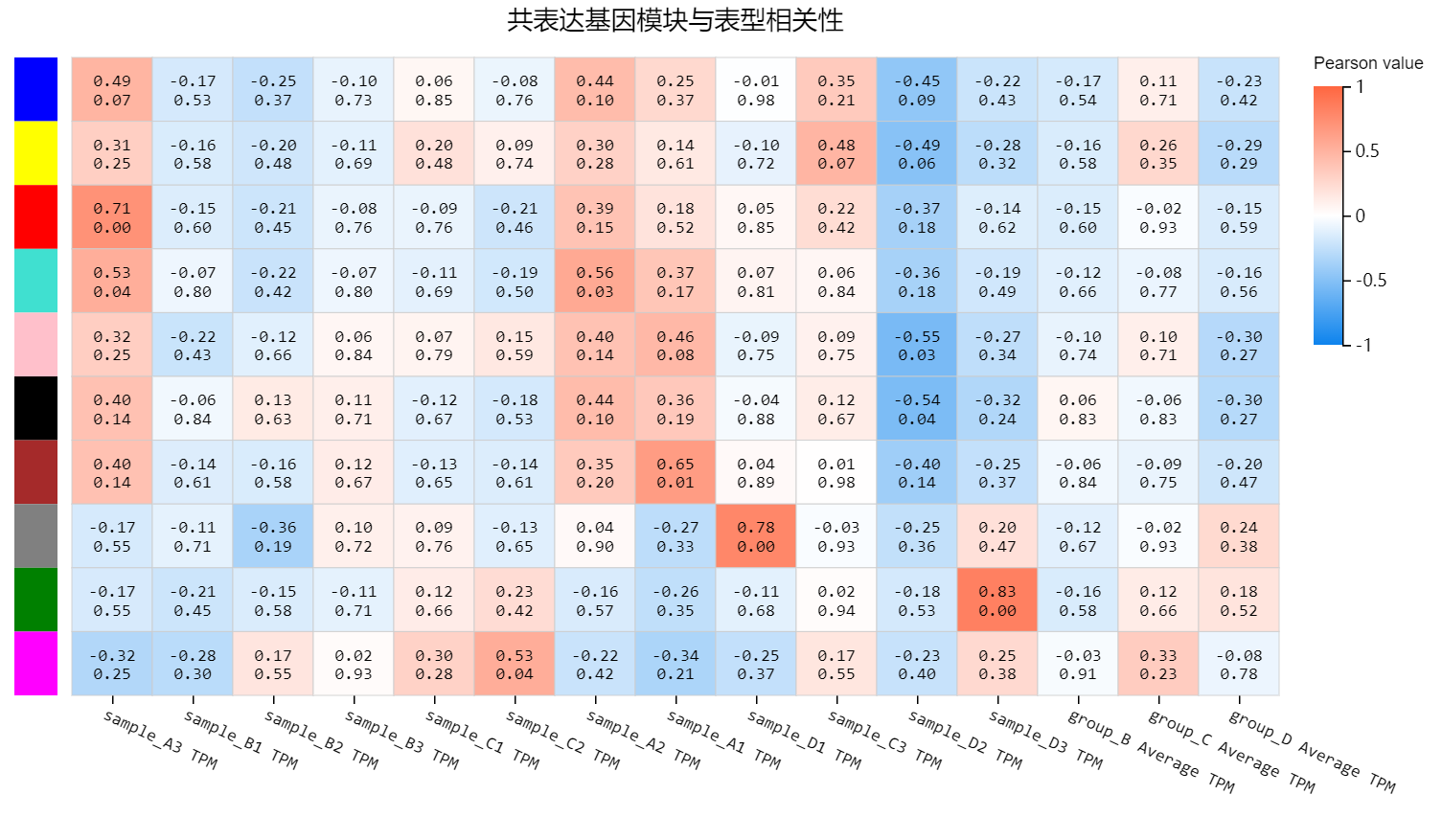
WGCNA
工具介绍:加权基因共表达网络分析,利用选定的基因 / 转录本 / 蛋白,构建共表达基因模块。并根据基因模块与样本(表型)的关联性与基因模块的内源性来鉴定关键基因。基于选定的基因 / 转录本 / 蛋白的表达量与样本(表型)相关性,作热图展示。官方推荐,至少需要 15 个样本以上的数据。
使用示例
系统仅展示共表达基因模块与表型相关性,详细结果可在页面上点击下载查看。
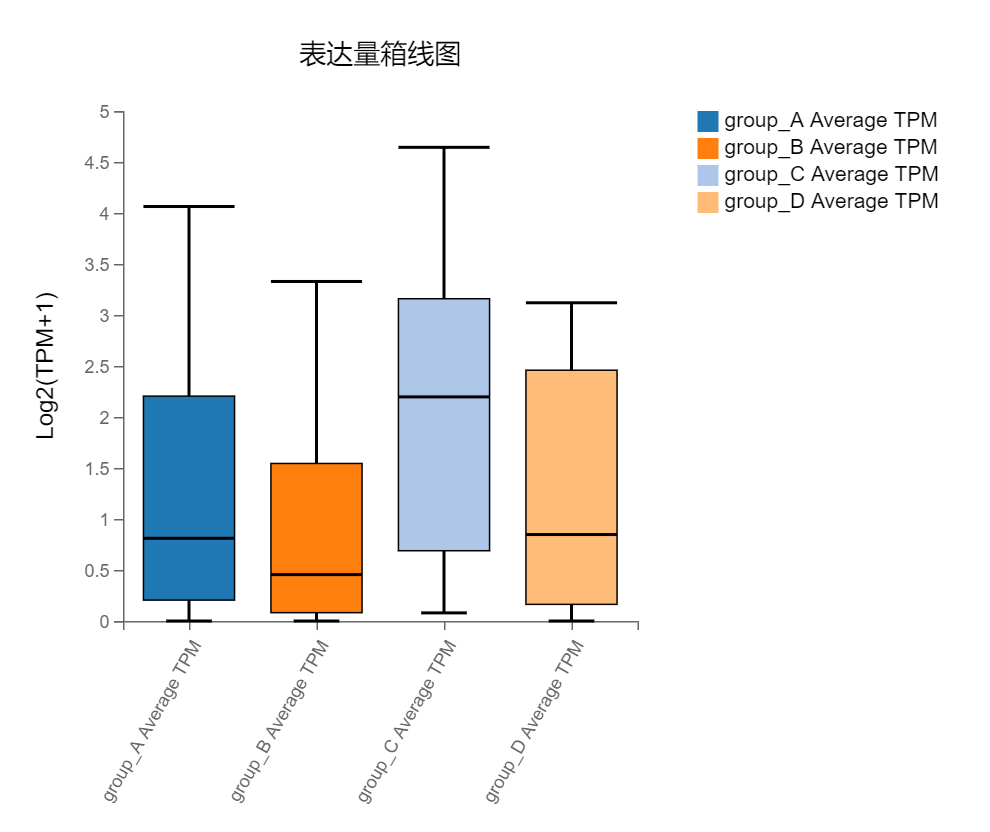
箱线图
工具介绍:选择指定样本,用表达量、差异倍数或自定义数据绘制箱线图。
使用示例
可切换展示箱线图和小提琴图。
PCA
工具介绍:主成分分析(Principal components analysis),对数据集进行降维,提取出数据集中对方差贡献最大的特征。对所有表达基因进行分析。
绘制方法:R 函数 princomp
使用示例
折线图
工具介绍:选定基因,使用表达量、差异倍数或自定义数据绘制折线图。
使用示例
火山图
工具介绍:根据差异比较方案,利用倍数变化(取log2)以散点的形式展示差异基因。
使用示例
散点图
工具介绍:选择两列数值型数据,绘制散点图,帮助了解其中的相关性。
使用示例
关联网络
工具介绍:对关联转换后的基因关系矩阵,绘制网络互作图,直观查看基因之间的互作关系。
参数解释 :
(详见对应 PPI\Targe\ceRNA\rnaplex\GGI 参数解释)
使用示例
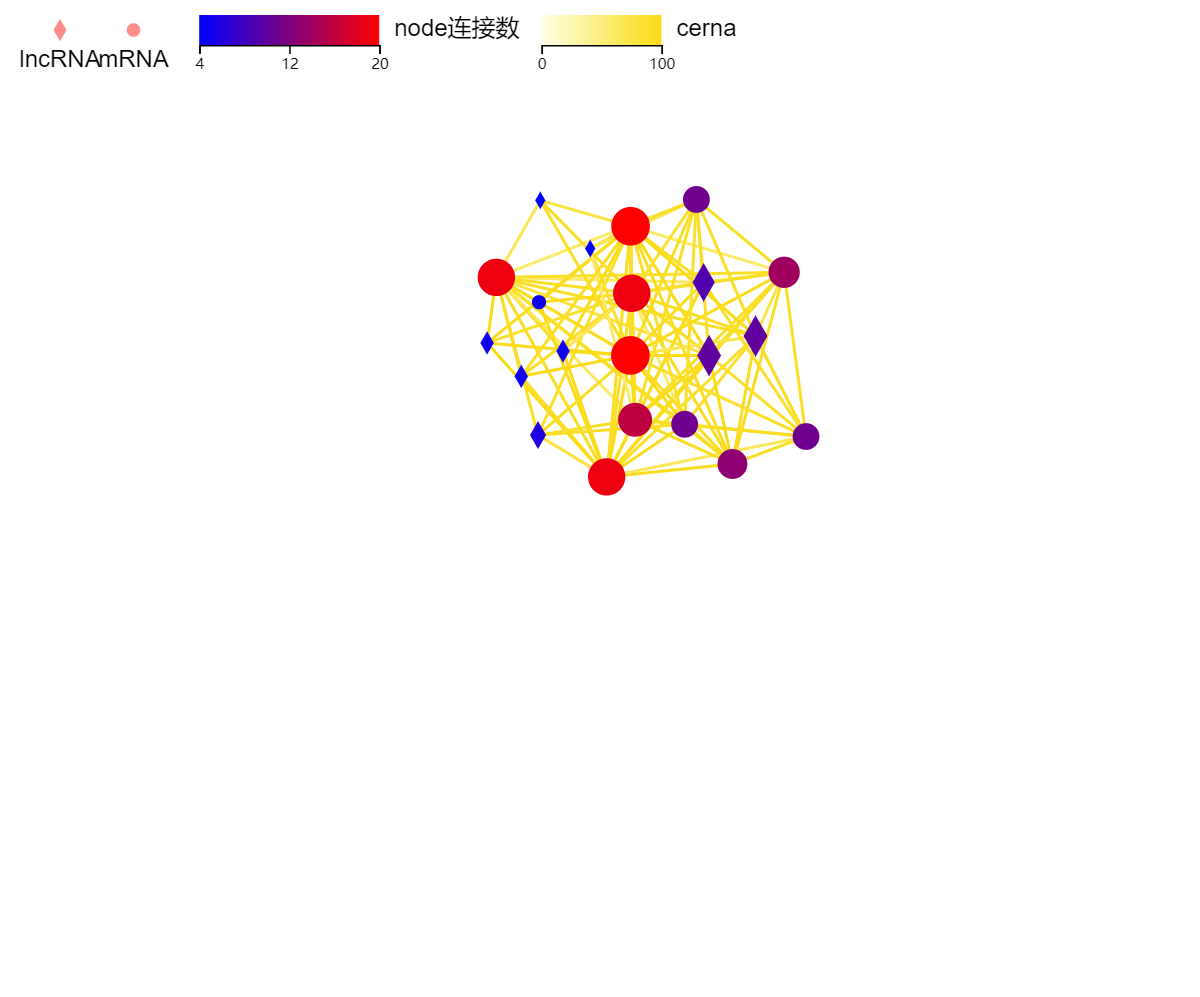
多组学关联
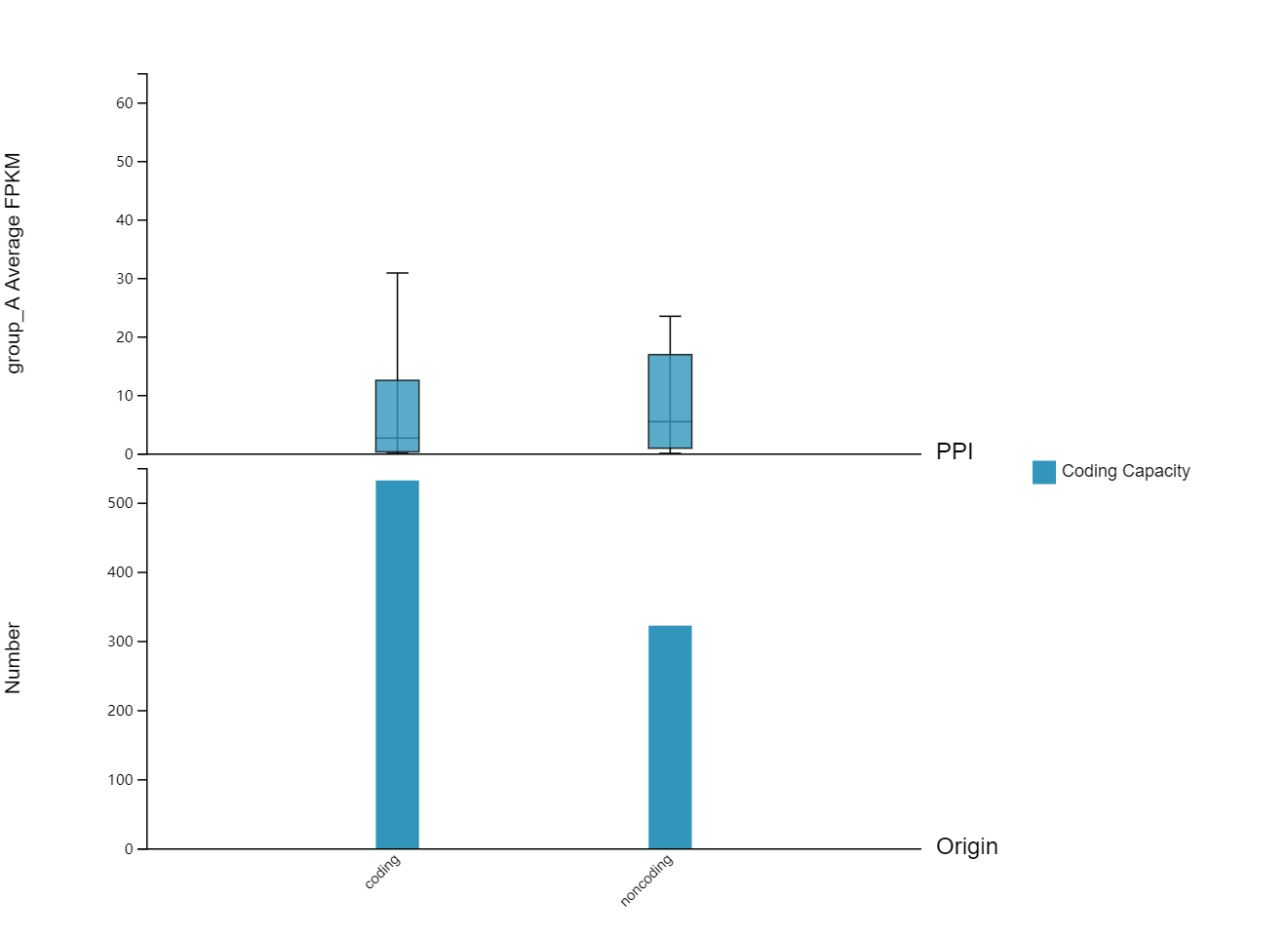
工具介绍:进行分类,绘制当前基因或通过 PPI、miRNA 靶标关系等扩展的基因的分类柱形图、表达量箱线图等。
使用示例
默认展示分类柱形图,可通过“设置”,添加所属分类的基因的表达量箱线图。
跨组学相关性
工具介绍:计算两组数据的相关性,例如miRNA与其靶基因在一组样本中表达量的相关性。
参数解释 :
各关系参数详见对应 PPI\Targe\ceRNA\rnaplex\GGI 参数解释
- 初始元件:选定的基因 / 转录本所在样本的表达量
- 关联元件:选定的基因 / 转录本经过关联转换后得到的基因所在的样本的表达量
使用示例
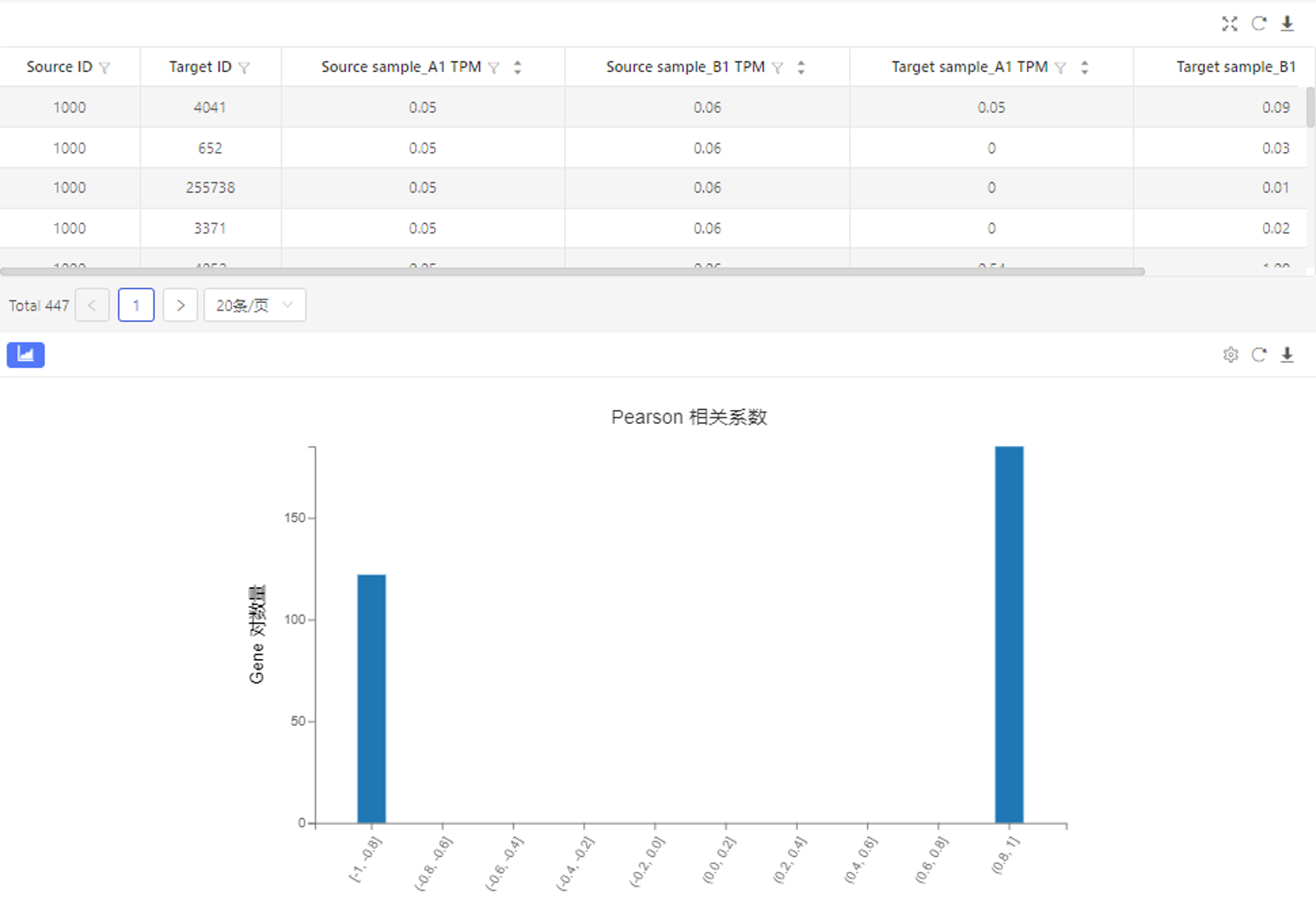 上方表格展示选定的初始元件与关联元件相关系数的具体数值。下方图片根据相关系数划分区间,只是不同区间内关联元件的个数。
上方表格展示选定的初始元件与关联元件相关系数的具体数值。下方图片根据相关系数划分区间,只是不同区间内关联元件的个数。
ceRNA
工具介绍:综合考虑mRNA/lncRNA与miRNA的靶向关系、RNA表达量相关性、超几何分布检验等,计算ceRNA关系,并绘制网络图
使用示例:
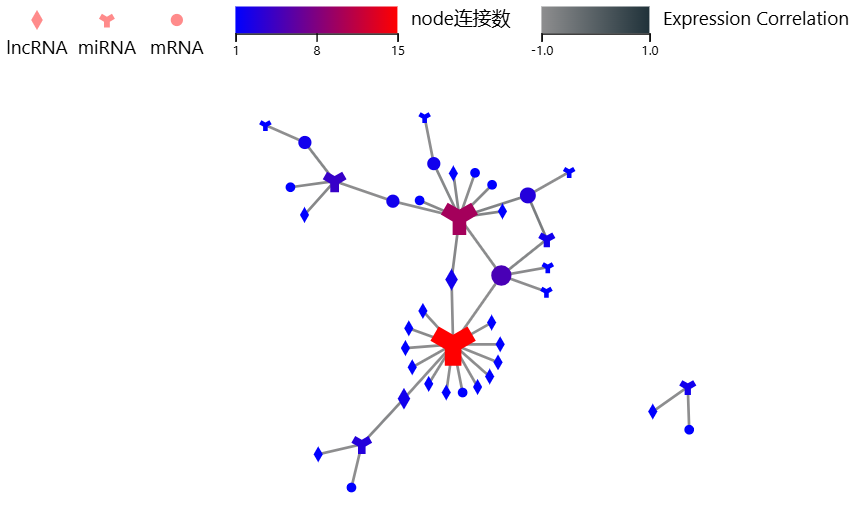
方法原理
根据用户设定的RNA表达量相关性及超几何分布检验P值的阈值,绘制ceRNA网络图。 参考该文献中的方法 Li Y, Jin X, Wang Z, Li L, Chen H, Lin X, Yi S, Zhang Y, Xu J. Systematic review of computational methods for identifying miRNA-mediated RNA-RNA crosstalk. Brief Bioinform. 2019 Jul 19;20(4):1193-1204. doi: 10.1093/bib/bbx137. PMID: 29077860.

通过R的phyper函数(https://stat.ethz.ch/R-manual/R-devel/library/stats/html/Hypergeometric.html)计算P值。
注意事项:该工具只考虑mRNA与lncRNA的ceRNA关系, 不支持circRNA,不支持mRNA-mRNA, lncRNA-lncRNA间的ceRNA关系
单细胞-差异分析
工具介绍:比较两个细胞集的基因表达情况,使用参数面板中选择的细胞集进行分析。可分析不同细胞类型/样本/自定义参数的差异基因表达情况。
使用示例
单细胞-细胞聚类
工具介绍:选择细胞进行聚类分析,分析内容包括细胞聚类、Marker 基因鉴定和细胞注释等。
分析软件:Seurat
细胞聚类降维方法:可选 UMAP 或 t-SNE 进行降维。与 t-SNE 相比 UMAP 更适合处理大型数据集和复杂的维度信息;结合了可视化的强大功能和减少数据维度的能力;除了保留本地结构外,它还保留了数据的全局结构。
Marker基因计算方法
- Wilcox:使用 Wilcoxon Rank Sum 鉴定 Marker 基因
- MAST:采用 hurdle 模型,用 MAST 包鉴定 Marker 基因
- LR:使用逻辑回归模型鉴定 Marker 基因
- poisson:使用泊松分布鉴定 Marker 基因。仅适用于含有 UMI 的数据集。
- negbinom:使用负二项广义线性模型鉴定 Marker 基因。仅适用于含有 UMI 的数据集。
- t:使用 t - 检验鉴定 Marker 基因
- bimod:使用似然比检验方法鉴定 Marker 基因
细胞注释数据库
CellMarkerCellMarker数据库为人类和小鼠各种组织的细胞类型提供了完整且精确的参考资源。该数据库对细胞标记信息、组织类型、细胞类型、癌症信息相关的十万余篇文献和4124条相关信息条目进行了人工校对,最终,整合了158种人类组织中的467种细胞类型,共13605个细胞标记基因,以及81种小鼠组织中的389种细胞类型,共9148个细胞标记基因。
Xinxin Zhang et al., CellMarker: a manually curated resource of cell markers in human and mouse, Nucleic Acids Research, Volume 47, Issue D1, 08 January 2019, Pages D721–D728
CancerSEA CancerSEA是首个从单细胞的角度解析癌症细胞不同功能状态的专用数据库,它提供了一个较为完整的癌症单细胞功能状态图谱:包含来自25种癌症的 41900个癌症细胞,共14种功能状态。
Huating Yuan et al., CancerSEA: a cancer single-cell state atlas, Nucleic Acids Research, Volume 47, Issue D1, 08 January 2019, Pages D900–D908.
细胞采样 当细胞数量较大时,Seurat支持对细胞进行采样(sketch)后再进行聚类。方法详见 https://satijalab.org/seurat/articles/seurat5_sketch_analysis
使用示例
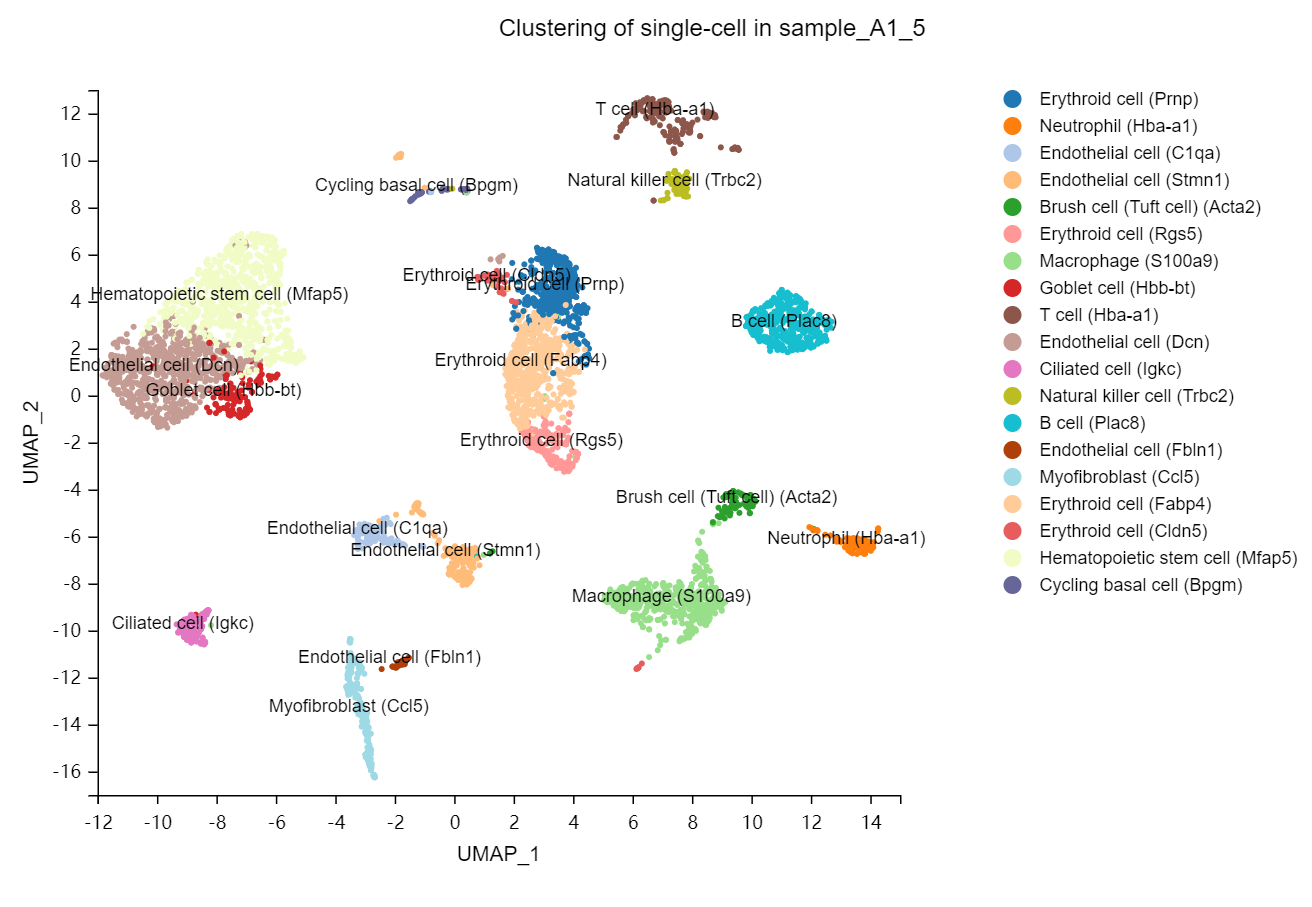
细胞聚类图:展示细胞聚类及注释情况。
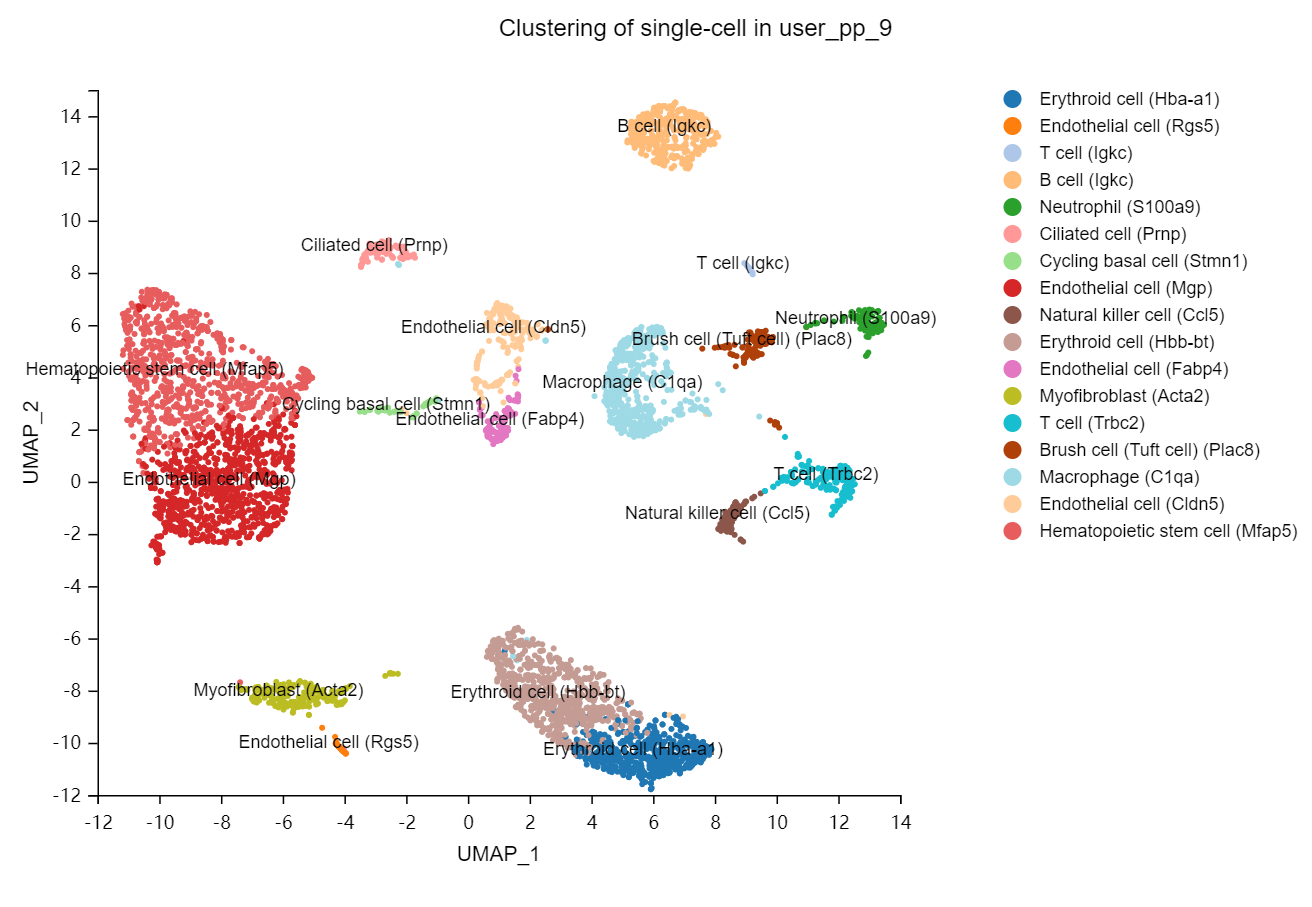
细胞基因数:展示该聚类细胞集中,各细胞类型的基因数统计。 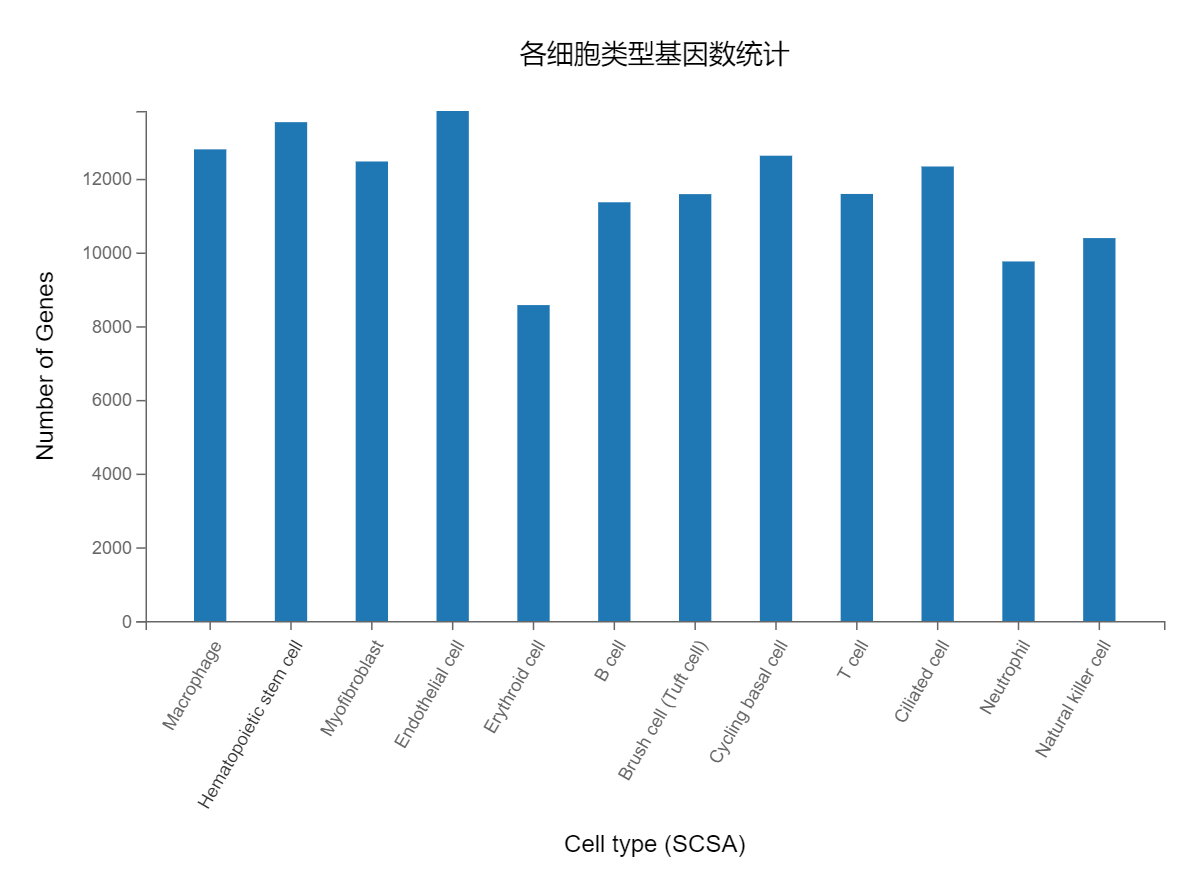
Marker基因:展示该聚类方案下,不同 cluster 的 marker 基因数统计。 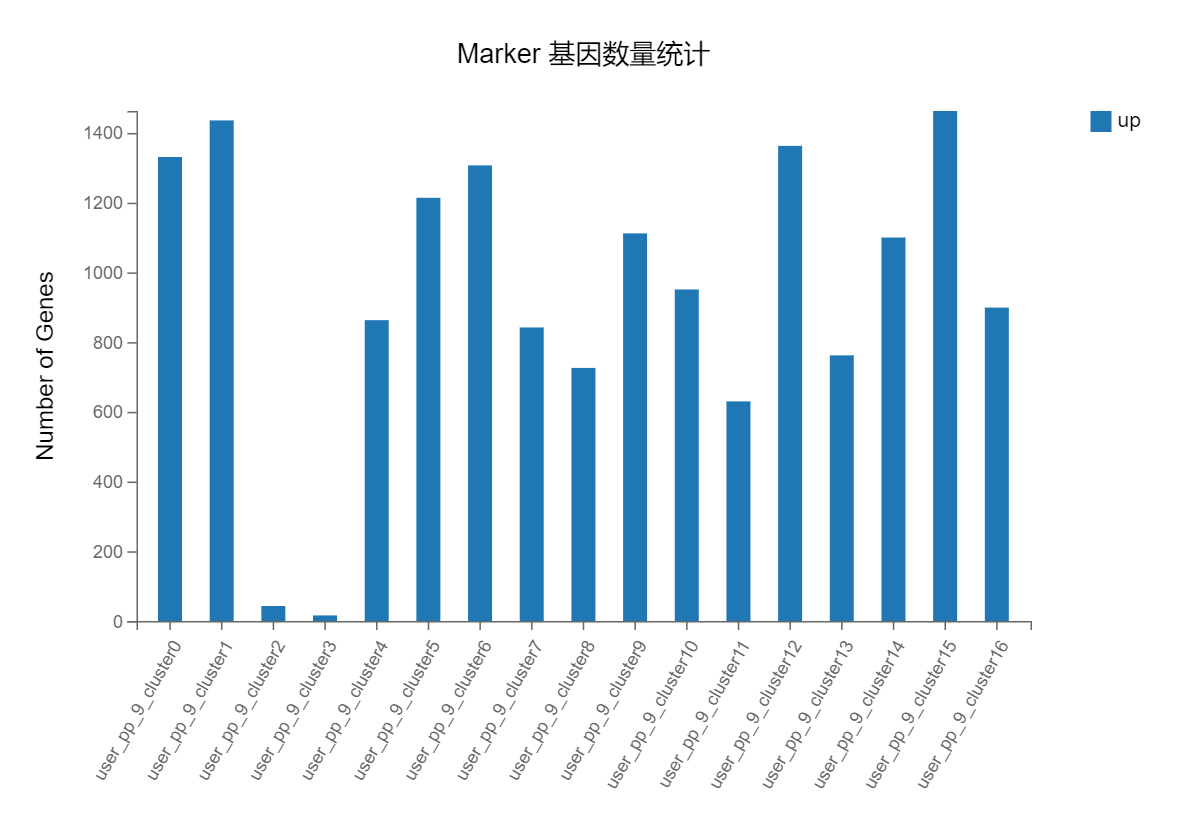
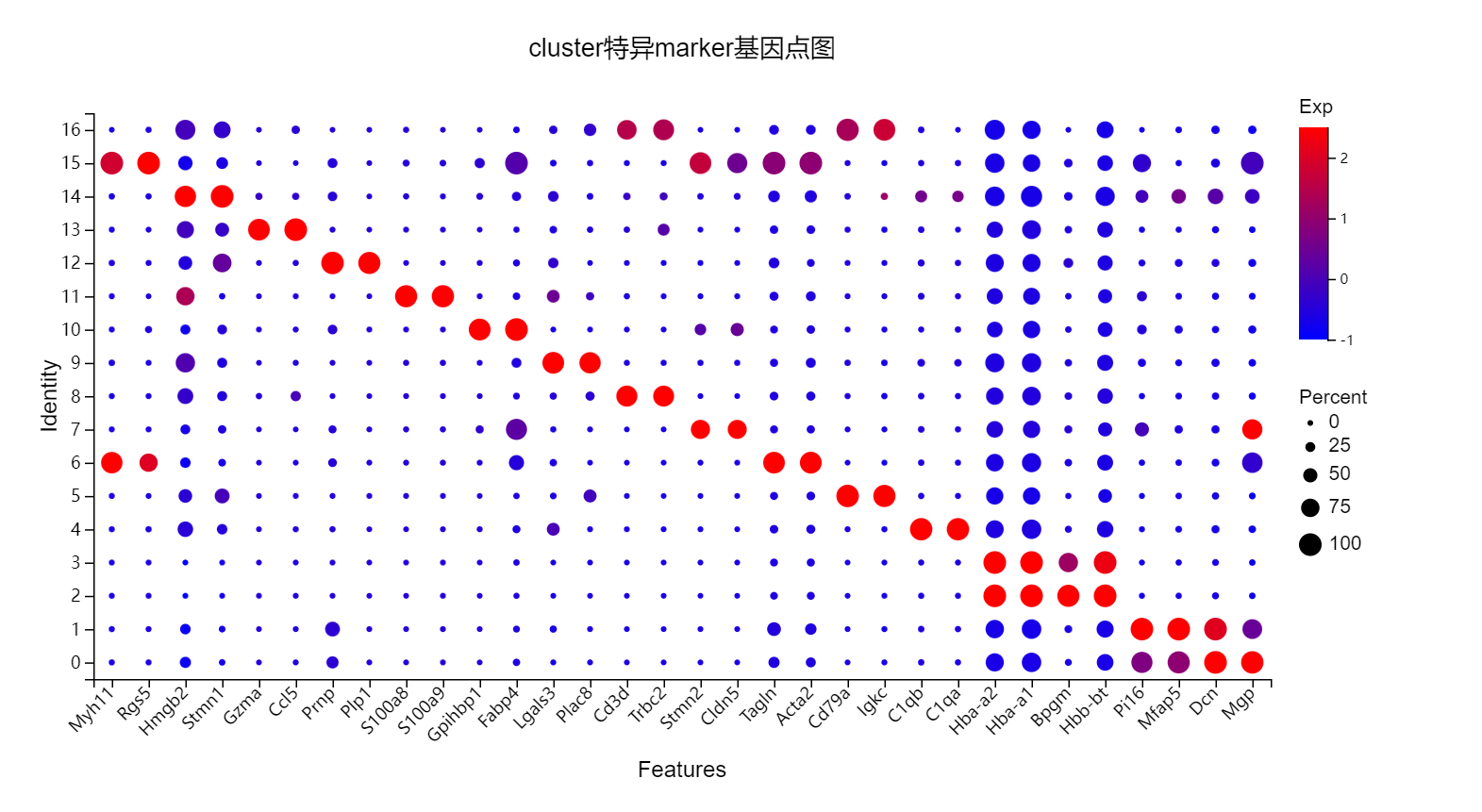
Marker基因表达特征:展示不同 cluster 中,最显著的前 2 个 Marker 基因的气泡图。
高变特征筛选:展示基因在聚类细胞集中的平均表达量及标准化方差,筛选高变基因。
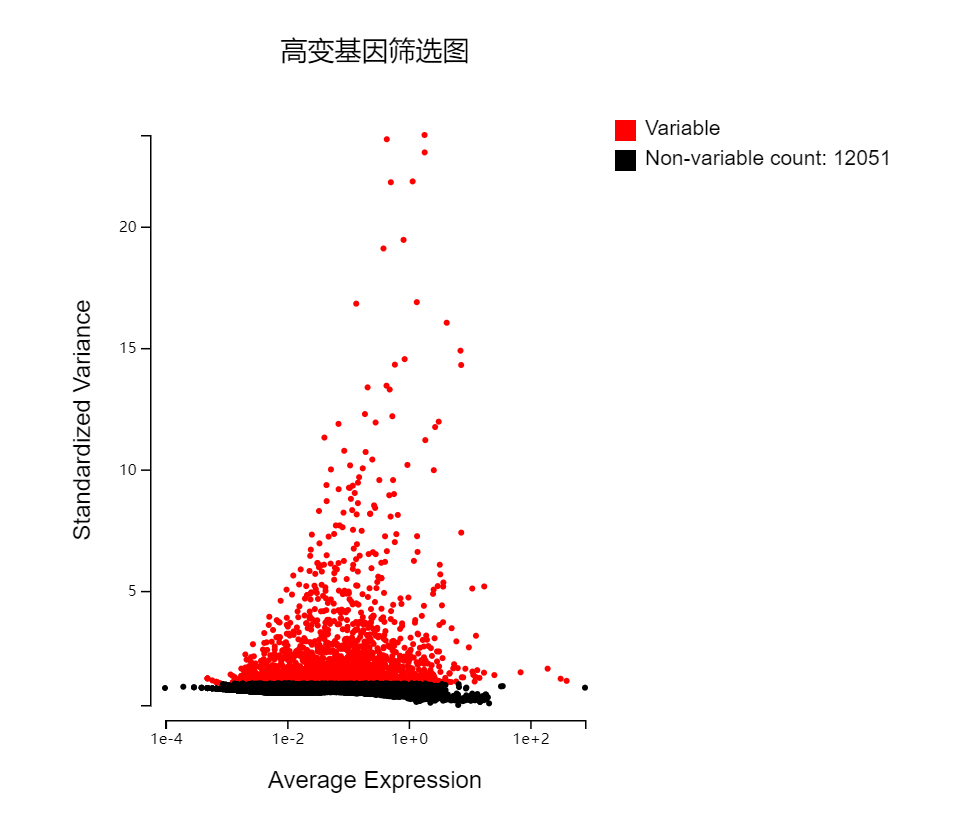
概况:描述该细胞聚类方案。

单细胞-细胞注释
工具介绍:对参数面板中选定的细胞进行注释,预测所属细胞类型。可选用 SCSA/SingleR/SciBet 分析软件,其中 SCSA 支持上传自定义数据库进行分析。
分析软件
SCSASCSA 是一种在 scRNA-seq 数据分析中,自动注释每个细胞 cluster 细胞类型的工具,该工具主要利用 cluster 中的差异表达基因(DEGs),以及这些基因所对应的细胞类型先验信息,从而对该 cluster 本身进行注释。该方法直接适用于 Seurat 所得到的标记基因筛选结果。为了消除标记基因的选择所带来的偏差,SCSA 将所得的所有标记基因与 CellMarker 和 CancerSEA 数据库中对应的信息整合为 “细胞类型矩阵”,通过一种打分注释模型来解析标记基因对应的细胞类型。除了已知的数据库,用户还可以根据相关领域的先验知识自定义标记基因与细胞类型之间的对应关系数据库。对于缺乏已知细胞标记的 cluster,该方法会对这些 cluster 中的标记基因进行 GO 分析,从而为用户提供研究的参考。
Cao Y et al (2020)., SCSA: A Cell Type Annotation Tool for Single-Cell RNA-seq Data. Front. Genet. 11:490.
SingleRSingleR 是一套对 scRNA-seq 数据进行无偏细胞注释的计算框架。该方法通过将单细胞转录组表达数据与参考数据集中纯细胞类型的微整列或 RNA-seq 数据进行对比,最终识别 scRNA-seq 数据中的细胞类型。
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M (2019). “Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage.” Nat. Immunol., 20, 163-172
SciBet SciBet 是一种基于标签数据对细胞类型进行监督注释的方法,该方法通过多项式分布模型和最大似然估计进行准确、快速的单细胞注释。
Li C, Liu B, Kang B, et al. SciBet as a portable and fast single cell type identifier[J]. Nature communications, 2020, 11(1): 1-8.
数据库
SCSA
CellMarkerCellMarker 数据库为人类和小鼠各种组织的细胞类型提供了完整且精确的参考资源。该数据库对细胞标记信息、组织类型、细胞类型、癌症信息相关的十万余篇文献和 4124 条相关信息条目进行了人工校对,最终,整合了 158 种人类组织中的 467 种细胞类型,共 13605 个细胞标记基因,以及 81 种小鼠组织中的 389 种细胞类型,共 9148 个细胞标记基因。
Xinxin Zhang et al., CellMarker: a manually curated resource of cell markers in human and mouse, Nucleic Acids Research, Volume 47, Issue D1, 08 January 2019, Pages D721–D728
CancerSEA CancerSEA 是首个从单细胞的角度解析癌症细胞不同功能状态的专用数据库,它提供了一个较为完整的癌症单细胞功能状态图谱:包含来自 25 种癌症的 41900 个癌症细胞,共 14 种功能状态。
Huating Yuan et al., CancerSEA: a cancer single-cell state atlas, Nucleic Acids Research, Volume 47, Issue D1, 08 January 2019, Pages D900–D908.
SingleR
共有 8 个数据库,其中 1-5 适用于人,6-7 适用于鼠。
- BlueprintEncodeData来自 Blueprint 和 ENCODE 数据库的 259 个 bulk RNA-seq 样本(Martens and Stunnenberg, 2013; The ENCODE Consortium, 2012)。其中,Blueprint Epigenomics 包含 144 个人类样本的纯免疫细胞 bulk RNA-seq 数据,共 28 种细胞类型;ENCODE 包含 115 个人类样本的纯基质细胞 bulk RNA-seq 数据,共 17 中细胞类型。
- DatabaseImmuneCellExpressionData来自 DICE 数据库的共 1561 个人类样本的纯免疫细胞 bulk RNA-seq 数据
- HumanPrimaryCellAtlasData来自人类原代细胞图谱(Human Primary Cell Atlas, HPCA)的共 713 个微阵列样本表达数据(Mabbott et al., 2013)。该数据集通过 Aran 等描述的方法进行预处理和归一化(Aran, Looney and Liu et al., 2019),每个样本注释 37 种主要细胞类型,157 种细胞亚型
- MonacoImmuneData来自 GSE107011 数据集共 114 个样本的分选免疫细胞 bulk RNA-seq 表达数据
- NovershternHematopoieticData包含 211 个人类样本的分选造血细胞微阵列表达数据
- ImmGenData来自 Immunologic Genome Project 的 830 个小鼠样本的纯免疫细胞微阵列表达数据(http://www.immgen.org/)
- MouseRNAseqData 包含 358 个小鼠样本的分选细胞 bulk RNA-seq 表达数据
SciBet 可选数据集
Scibet - 共有 90 + 数据集可供选择,点击 查看列表
使用示例
单细胞-拟时间分析
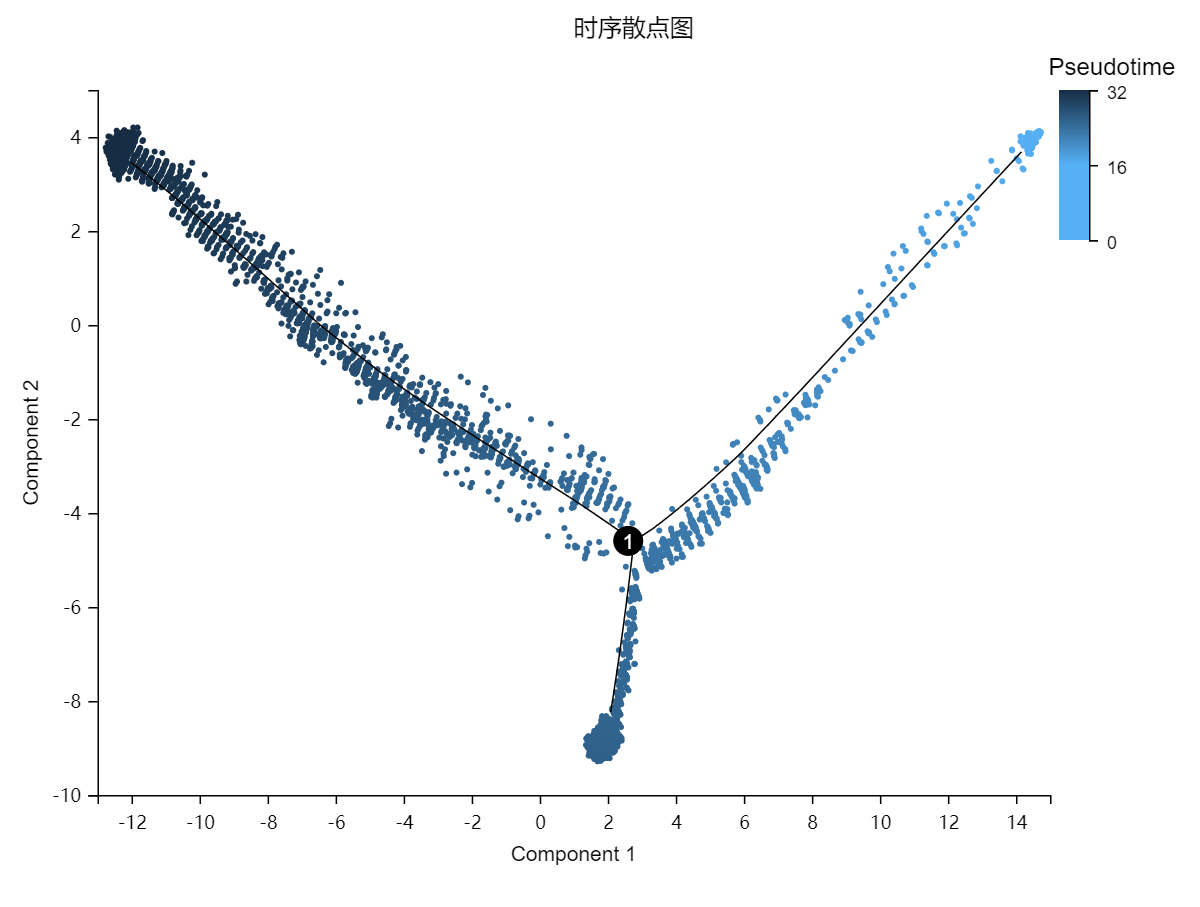
工具介绍:对选定细胞集进行拟时间分析,通过分析每个细胞中关键基因的表达模式,将相关细胞按照一定顺序排布在拟时间轴上,模拟发育过程中的细胞分化过程。
使用示例
拟时间概览:展示所选细胞的拟时间分析结果。
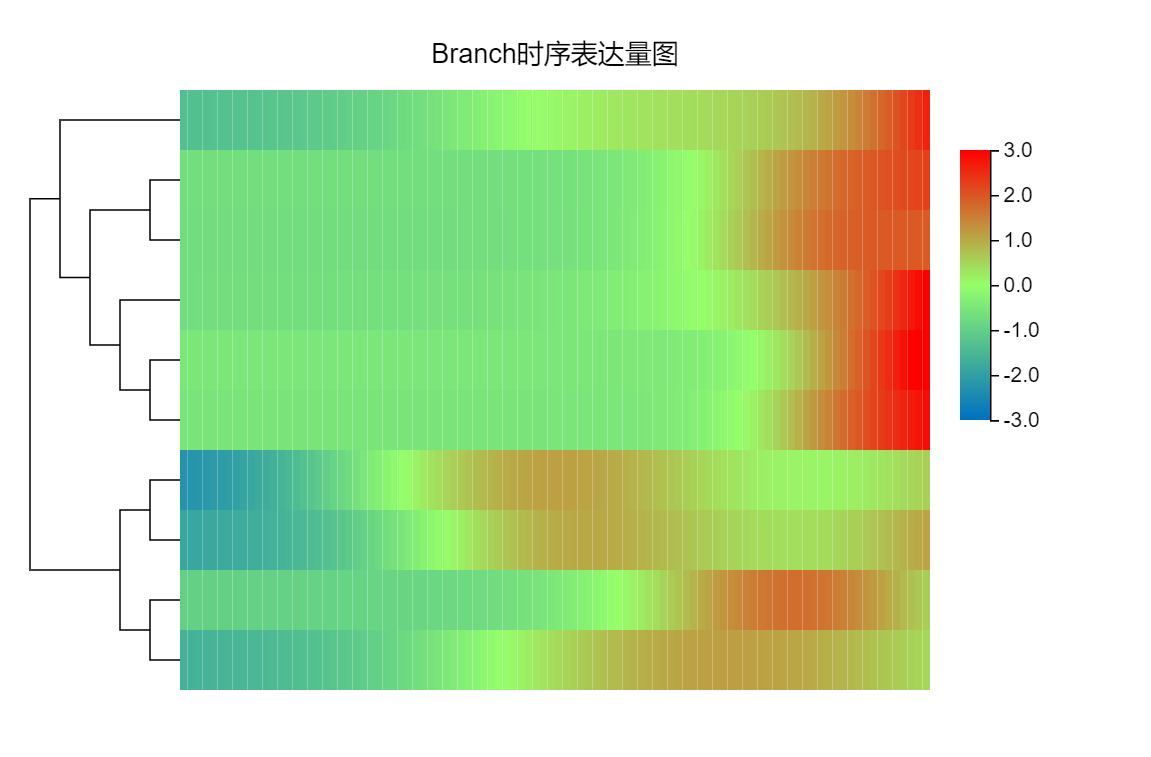
Branch表达量热图:展示不同的拟时间点上,各比较组差异基因的表达情况。
基因表达量散点图:展示不同基因在各拟时间点上对应的表达量散点图。
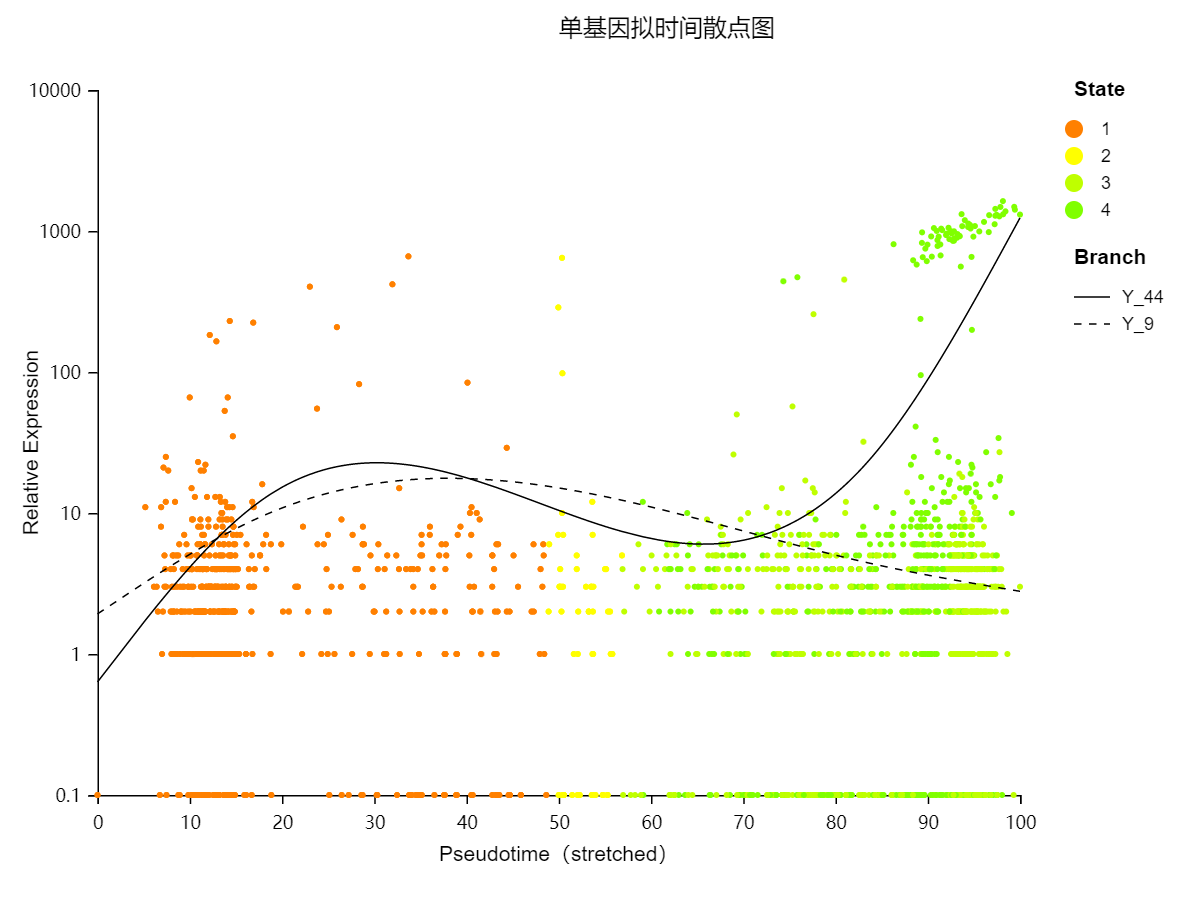
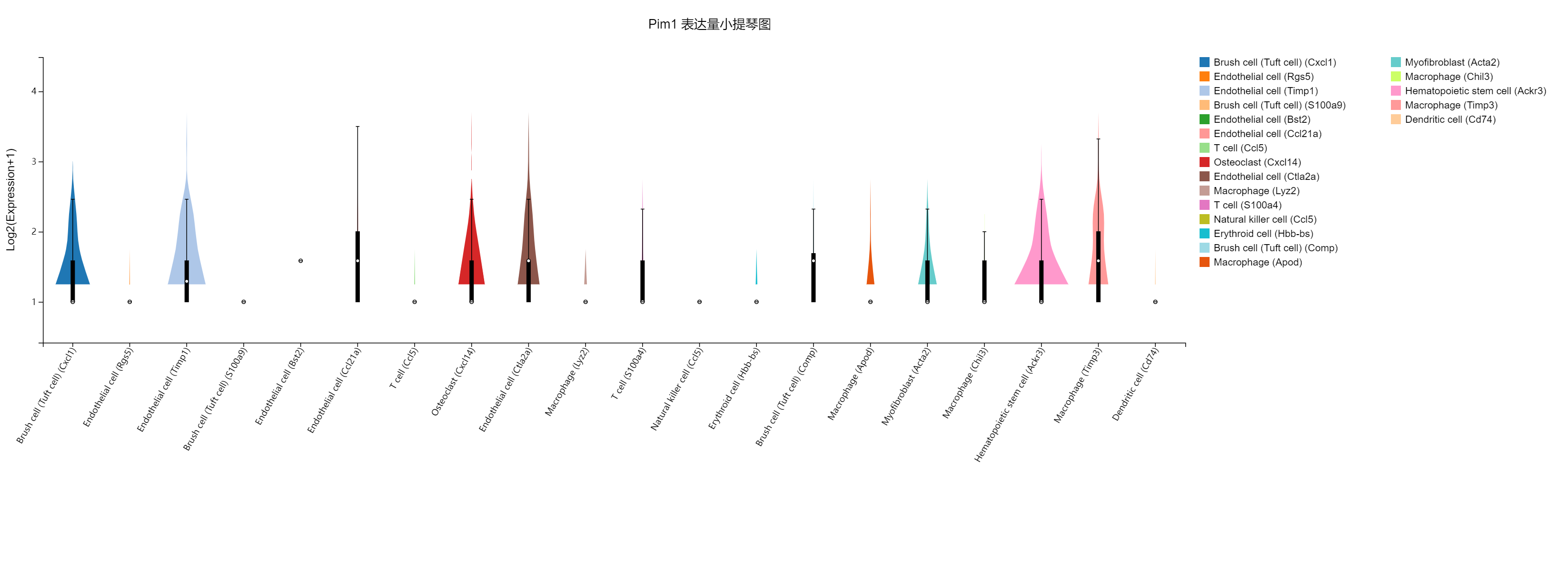
单细胞-箱线图
工具介绍:在当前页面的表格或聚类图中选择细胞,用箱线图 / 小提琴图展示在不同的样本 /cluster/ 细胞类型 / 拟时间点的基因表达量 / UMI 数量。
使用示例
可切换箱线图 / 小提琴图展示。