宏基因组项目
宏基因组项目简介
微生物群 (Microbiota) 被定义为占据特定环境的微生物的集合,包括细菌、真菌、古菌和病毒。从自然界中分离出微生物进行纯培养是最基本、最常用的微生物研究方法。然而,据估计自然界中超过 99% 的微生物不能通过传统的分离培养技术获得其纯培养,因此大量环境微生物不能被研究和开发利用。此外,随着微生物活性产物的广泛研究和深入开发利用,从环境微生物中分离出新的菌株以及筛选出新活性物质的几率也受到限制,如何发现并利用未知微生物成为微生物研究的重要课题之一。随着高通量测序 ( High-throughput sequencing,HTS)或下一代测序(next generation sequencing,NGS) 技术的发展,微生物研究开始采用扩增子测序 (Amplicon sequencing) 和宏基因组测序 (Metagenomics sequencing) 等基于测序的方法。这些测序方法使得研究群落中不可培养的微生物成为可能。
通过测序获得环境样品中的所有微生物遗传信息,再通过生物信息学分析获得目标微生物群落组成和功能等信息,使得对微生物的研究可以绕开微生物的分离培养过程,从而提供了一种对不可分离培养的微生物进行研究的途径。
宏基因组(Metagenomics)项目基于高通量测序的序列信息,首先进行质控、序列组装和基因预测获得基因信息,再将基因注释到物种和功能数据库,可以了解环境样品中的微生物的物种分类、群落结构、基因功能以及代谢网络等微生物群落组成相关信息,通过统计学分析方法可以比较不同样品之间物种组成、群落功能的差异。
基于高通量测序的微生物组学研究
- 确定研究目标并设计研究方案 按照研究的目的有两种类型的宏基因组研究类型。A) 一种是为了探究不同处理,不同时空环境样本的宏基因组的比较研究,如比较不同施肥处理大豆根际微生物组成的差异,比较疾病组与对照组肠道微生物群落差异,确定生物标志物。这类研究要求至少有 2 个分组,每个分组至少需要 3 个生物学重复,对于肠道等个体差异较大的样本推荐生物学重复数量大于 5。试验前设定好采样计划,按照采样点进行采样,如果需要进行与环境因子的关联分析,环境因子的的重复属需要和测序样本数量对应。B) 另外一种是探究罕见样本中的微生物组成研究,如深海沉积物,火山口土壤,月球土壤等样品,宏基因组联合其他组学可以从基因组层面、转录表达层面以及代谢层面研究环境样本中微生物群落的功能。
- 执行研究方案并采样 按照研究方案严格执行实验,并在实验结束时,按照计划在无菌环境下采集对应的样本,根据样品类型选择合适手段送到实验室,并尽快完成样本 DNA 提取。
- 提取样本 DNA 通常环境样本的样本量比较少,所以通常会使用试剂盒提取,提取完的 DNA 应该尽快使用干冰运送到华大测序实验室。同时,华大也提供了 DNA 提取服务。
- 测序流程 DNBSEQ 平台采用 RCA(Rolling Circle Amplification, 滚环扩增)。RCA 测序每次扩增都使用第一次的模板进行线性扩增,避免了测序过程中的错误累积,具有更低的扩增偏差和更高的准确性。
- 生信分析 按照实验设计,确定分析方案,对下机序列进行生物信息学分析并将结果可视化。
建库与测序流程
DNA 在建库前会先进行质检,检验其浓度 (≥ 12.5 ug/ul)、完整性(电泳胶图主峰 > 20 kb)和纯度(无蛋白、RNA / 盐离子污染),达到条件的样本会进入建库流程。文库构建的具体流程如下:
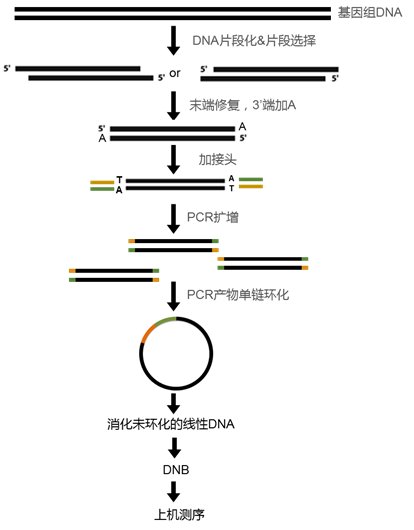
- 样品打断。取一定量的宏基因组 DNA ,用 Covaris 超声波破碎仪将其打断。
- 片段大小选择。打断后的样品磁珠进行片段选择,使得样品条带集中在 200-400bp 左右。
- 末端修复、加 “A”、接头连接。配制反应体系,适温反应一定时间,修复双链 cDNA 末端,并在 3’ 末端加上 A 碱基,配制接头连接反应体系,适温反应一定时间,使接头与 DNA 连接。
- PCR 反应及产物回收。配制 PCR 反应体系,并设置反应程序,对连接产物进行扩增。扩增产物用磁珠进行产物纯化回收。
- 产物环化。将 PCR 产物变性为单链后,配制环化反应体系,充分混匀适温反应一定时间,得到单链环形产物,消化掉未被环化的线性 DNA 分子后,即得到最终的文库。
- 文库检测。环化产物进行上机前浓度检测。
上机测序
检测合格的文库将会被安排上机测序(DNBSEQ):单链环状 DNA 分子通过滚环复制,形成一个包含多个拷贝的 DNA 纳米球(DNB)。将得到的 DNBs 采用高密度 DNA 纳米芯片技术,加到芯片上的网状小孔内,通过联合探针锚定聚合技术(cPAS)进行测序。宏基因组采用 MGISEQ 2000 平台(或 DNBSEQ T7),PE150 测序。
生信分析流程
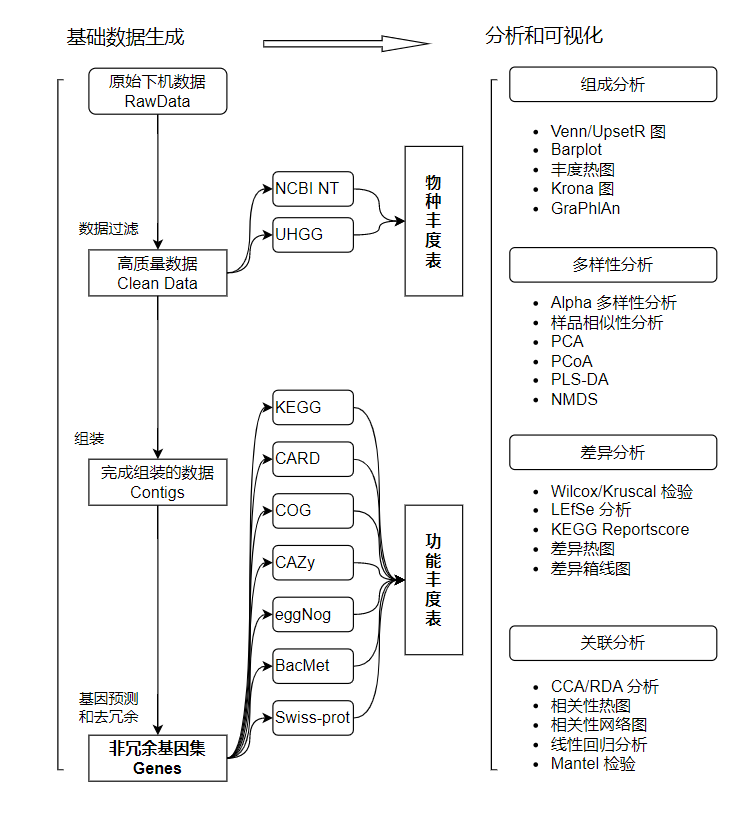
- read 质控和去宿主:使用 SOAPnuke 软件对原始数据进行过滤质控,使用 Bowtie2 比对宿主序列并去除比中的序列,生成 Clean Data。
- 功能注释:先使用 MEGAHIT 对完成质控和去宿主的序列进行基于 k-mer 的组装,生成 contigs,再用 MetaGeneMark 软件,预测 contigs 中的基因序列。使用 CD-HIT 软件,对得到的基因进行去冗余,使用 Salmon 软件统计各个基因相对丰度表,再使用 DIAMOND 或 RGI 将非冗余基因比对到 eggNOG、KEGG、BacMet、CARD、COG、CAZy、Swiss-prot 等数据库,完成基因功能注释。
- 物种注释:使用 Kraken2 和自建数据库(筛选 NCBI NT 数据库或 UHGG 数据库)比对来计算样品中所含有物种的序列数,再用 Bracken2 来对样品中物种的实际丰度进行估计,完成物种注释。
- 基于基因丰度表、物种丰度表和功能丰度表,可以对基因、物种和功能分布情况进行可视化,计算其 Alpha 多样性、Beta 多样性,进行物种或功能丰度聚类分析,PCA、PLSDA、PCoA 和 NMDS 降维分析,样品聚类分析,使用 Wilcoxon/Kruscal,T 检验/方差分析、LEfSe 分析以及 KEGG 通路富集分析(Reportscore 方法)挖掘样品之间的物种组成和功能组成差异。
- 结合环境因子、病理指标或特殊表型进行深入关联研究,能够为进一步深入研究和利用样品的物种和功能提供理论依据。