物种注释
简介
宏基因组探究样品中微生物的组成时,将 Clean Data 中每一条 read 比对到数据库获得该 read 对应的物种信息,所有的 read 注释结果就构成了群落的组成信息,再通过算法构建物种的丰度,得到物种丰度表。
Kraken 物种注释
Kraken2 [1][2] 是一个基于 k-mer 物种注释软件,相比于传统的序列(或基因)比对(或机器学习)方法,Kraken2 具有速度更快和对碱基差异更敏感的优点,其原理如下:
mer 是 monomeric unit,一个 mer 可以看成是一个碱基,k-mer 指的是含有 k 个碱基的核苷酸序列。Kraken2 首先会按照要求,将一个或多个来源的注释数据库构建一个子库,并将所有可能物种的基因组序列拆分成 k-mer 存储。对于任意需要比对的长度为 n 的未知序列,首先被拆分成 n - k +1 个 k-mer ,然后逐个将这些 k-mer 和预先构建的 k-mer 数据库进行比对,所有命中数据库的 k-mer 会被放置在一个分类树上,未命中的 k-mer 会被忽略,分类树上节点记录了命中 k-mer 的数量,然后根据最低共同祖先(LCA)算法,选择分析获得分类树中连续的最低共同祖先。
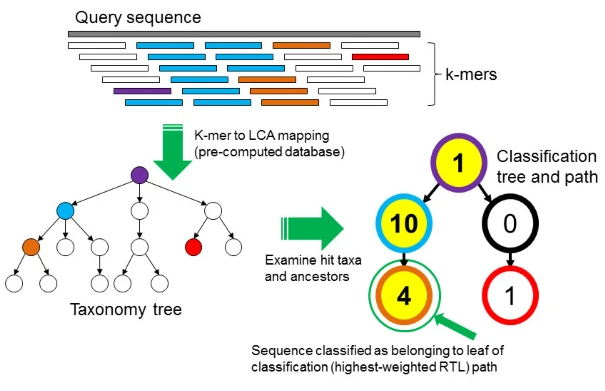
图1:Kraken2 算法原理。
第一步,将待预测的序列打断成 k-mer ,图中的小片段;
第二步:根据LCA算法将所有的 k-mer 放置在分类树的结点上,每个节点的数字表示命中该节点的 k-mer 数量,可以看到分类树上命中的节点连接起来可以组成不同的路径;
第三步,统计每个路径上命中 k-mer 数量之和,称为得分;
第四步,得分最高的路径所在的分类即为该序列的物种注释结果,如果两个最长路径得分相同采用 LCA 算法取他们的最低公共祖先。
Kraken2 进行物种注释需要比对数据库,Dr.Tom 提供了两个注释用数据库,并根据样本来源自动选择合适的数据库:
| 数据库 | 构建方式 | 版本 |
|---|---|---|
| UHGG [3] | 官方构建 Kraken2 数据库 | v2.0 |
| NCBI NT [4] | Kraken2 官方预先构建子库 | k2_pluspf_16gb_20240112 |
按照样本来源选择合适的数据库:
人肠道样本:采用人类胃肠道基因组(Unified Human Gastrointestinal Genome, UHGG) 数据库,该数据库收录了 20 多万种人类肠道微生物基因组。
其他样本:采用根据 NCBI NT 数据库 构建的子库,NCBI NT 数据库收集了来自 GeneBank、RefSeq、TPA 和 PDB 等数据库的基因组,基因,转录序列等数据,可用于生物信息数据的发掘。但因其数据过于庞大,因此在实际分析过程中,会构建一个子库,使用子库进行物种注释。
| 软件 | 版本 | 默认命令 |
|---|---|---|
| Kraken2 [1:1] | 2.1.2 | kraken2 --paired --use-names --gzip-compressed |
Bracken 物种丰度构建
Kraken2 将每个 read 进行物种分类和注释,但并非每个 read 都能注释到种水平,有些 read 只能注释到属甚至更高水平,如果只是简单根据 read 和物种注释的结果,进行简单的加和统计物种丰度的结果是不准确的。Bracken2 [5] (Bayesian Reestimation of Abundance after Classification with KrakEN) 基于贝叶斯定理将 Kraken 注释结果中无法注释到低水平的更高水平分类以概率估计 (probabilistic estimate) 的方式分配 (re-assign) 到低水平中;对于亚种水平的分类,Bracken2 直接将其累加到种水平。
通过上述的计算方式,Bracken 实现了更精确的丰度构建。得到的丰度表将用于后续的统计学分析。
| 软件 | 版本 | 默认命令 |
|---|---|---|
| Bracken2 [5:1] | 2.6.1 | bracken -r 150 -l S 注意:r 表示读长,取值为 100 或者 150 |
相关信息
在 Clean Data 中,物种丰度表中的样品名是送样时的名称或二次确认的样品名,路径为
- TaxonomyAnalysis/Abundance/{分类水平}.relative.xls:指定分类水平的相对丰度表
- TaxonomyAnalysis/Abundance/{分类水平}.absolute.xls:指定分类水平的绝对丰度表
也可以在在线报告系统顶部的下载分析结果下载丰度数据,此时样品名是填写方案时的样品名,路径为
- TaxonomyAnalysis/Rename_Abundance/{分类水平}.relative.xls:指定分类水平的相对丰度表
- TaxonomyAnalysis/Rename_Abundance/{分类水平}.absolute.xls:指定分类水平的绝对丰度表
常见问题
Q:注释后物种丰度数据在哪里下载?
A:进入系统,点击页面顶部的下载分析结果按钮,您可以下载所有的分析结果,解压后您可以看到完整的物种丰度数据。
路径为:TaxonomyAnalysis/Rename_Abundance,其中:
- {分类水平}.absolute.xls 为 指定分类水平的绝对丰度表
- {分类水平}.relative.xls 为 指定分类水平的相对丰度表
- {分类水平}.{数据类型}.xls.header 为分析临时数据,不用管他
Q:为什么有的项目注释数量物种数目多,有的少?
A:物种注释结果数量大小与以下 3 点有关:
- 环境的复杂程度:环境越复杂,注释物种越多,反之亦然。土壤、水体等环境属于高复杂度环境,而肠道(粪便)样本属于低复杂度环境。
- 样本数量:随着样本数增加,样本中独有的物种被检测出来,会增加物种注释的总体数量。
- 测序深度:在测序数据量越高,越能把样品中稀有的物种注释出来。
若您测序结果中注释得到的物种很少,排除实验(实验污染)及测序(数据量)的问题,可能有以下原因:
- 所测的环境样品自身属于低复杂环境,含的物种较少
- 所送的样本组数太少,无法覆盖绝大部分物种
- 所关注的物种研究较少,数据库中物种的代表序列及相关信息较少
Q:注释结果中 NA 是什么意思?
A:宏基因组项目物种注释采用的是 Kraken2 软件,其原理是将测序序列打断成长度为 k(默认k=35)的子序列,称为 k-mer,然后将 k-mer 比对到自建数据库。比中数据库的 k-mer 会被分配一个到物种分类树上,没有比中的会被忽略,当一条序列的所有 k-mer 都有注释结果,再按照规则从注释树上选择最低层的物种注释结果即为这条序列的物种注释结果。但所有的 k-mer 可能只被注释到高层级,如属、科、纲,此时该序列的低层级会被标记为 NA。
Q:物种注释用数据库选择依据?
A:系统自动根据样本类型选择物种注释用数据库,若样本来源人肠道则选择 UHGG 数据,其他来源则选择 NCBI NT 数据库。
参考文献
Wood, D. E., & Salzberg, S. L. (2014). Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biology, 15(3), R46. https://doi.org/10.1186/gb-2014-15-3-r46 ↩︎ ↩︎
Wood, D. E., Lu, J., & Langmead, B. (2019). Improved Metagenomic Analysis with Kraken 2. Genome Biology, 20(1), 257. https://doi.org/10.1186/s13059-019-1891-0 ↩︎
Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., Pollard, K. S., Sakharova, E., Parks, D. H., Hugenholtz, P., Segata, N., Kyrpides, N. C., & Finn, R. D. (2021). A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome. Nature Biotechnology, 39(1), 105–114. https://doi.org/10.1038/s41587-020-0603-3 ↩︎
Home—Nucleotide—NCBI. (n.d.). Retrieved May 6, 2022, from https://www.ncbi.nlm.nih.gov/nucleotide/ ↩︎
Lu, J., Breitwieser, F. P., Thielen, P., & Salzberg, S. L. (2017). Bracken: Estimating Species Abundance in Metagenomics Data. PeerJ Computer Science, 3, e104. https://doi.org/10.7717/peerj-cs.104 ↩︎ ↩︎